![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
188 Cards in this Set
- Front
- Back
|
liver and glucose homeostasis |
key organ - main site of gluconeogensis ------ receives glucose from intestine via the portal vein |
|
|
fates of glucose |
can either go to pyruvate via glycolysis or to glycogen in the liver or muscle |
|
|
glycogen breakdown |
via glycogenolysis and it turns into energy and pyruvate |
|
|
gluconeogenesis |
supported by aa and by TAG but not by FA |
|
|
brain's response to low glucose |
--pititary gland - releases cortisol ----------- adrenal medulla - releases epi --> makes glucose ------ sympathetic system - releases norepi and ACh --> increases fat and muscle increase production and uptake . muscle stimulates breakdown of aa |
|
|
gluconeogenesis |
in liver - coordinated by muscle(lactate and aa) and adipose ( tag --> fa and glycerol) |
|
|
hierarchy of response to low glucose |
1) insulin decreases 2) glucagon increases 3) epinephrine increases - critical when glucagon is gone but not as effecient 4) cortisol adn growth hormones - adapations to prolonged fasting and insulin resistin |
|
|
insulin biosynthesis |
in beta cells in central part of islet cells arterial blood can enter so it can sense directly how much glucose is in the blood---- uptake facilitated by GLUT 2 -------- insulin is a pepetide hormone and its amino terminal gets cleaved along with some other parts in the golgi adn then there are two peptide chains joined by a disulfide bond ------------- c peptides are also released when insulin is made - this is how you can test how much de novo insulin is in the system ---------------- they also secrete islet amyloid polypeptides that can make up amyloid fibrils that aggregate |
|
|
mechanism of insulin secretion |
glucose enters via glut 2 and this goes to pyruvate (into TCA cycle) --> increase ATP --> closes K+ channels --> depolarization --> opens Ca2+ channels --> fusion of vescicles that lead to insulin secretion |
|
|
uncoupling protiens |
idk what is but if you increase them you decrease insulin secretion |
|
|
sirt and insulin secretion |
sirt inhibits PCP1 which causes a faster closing of the K+ channels which leads to more secretion |
|
|
c peptide |
released with insulin, good for testing how much insulin is in the blood because insulin is degraded fast but this isnt |
|
|
other factors that affect insulin secretion |
AC, PLC, activate cAMP or insitol pathway |
|
|
glucose trasnporters |
SGLT 1/2 - in intestine and kidney - active transporter GLUT 1 - in brain GLUT 4 - insulin sesnsitive, translocated in muscle and fat when high glucose GLT 2 - no affinity but high capacity - in beta cells and liver |
|
|
canonical insulin signalling |
akt/pkb and mek ---> insulin receptor and IGFR regulate metabolism - activation of insulin receptor can chane transcription for many processses |
|
|
lipid metabolism |
insulin promotes TG storage and mobilization by increase LPL and FA uptake, inhibit HSL and increasing lipogensis |
|
|
obesity |
a measure through BMI - caused by energy imbalance but can be influences by genetics and monogenetic mutations - potentially other thigns as well like sleeping, smoking, prenatal effects, pollution |
|
|
BMi |
body weight (kg) / body height squared (m) |
|
|
what are the different forms of diabeted insipidus and what is the common traits |
1) central 2) nephrogenic 3) dipsogenic 4) gestational - all diabetes insipidus has polyuria (excessive pee) and polydipsia (excessive thirst) |
|
|
cenetral diabetes insipidus |
most common form - defeciency or resistance to vasopression/adh. the brain can sense the amount of water via hormones in the hypothalamus. something can go wrong in the hormones or in the kidney |
|
|
vasopressin |
regulates aquaporins in the kidney which regulates water trasnport. increases when there is high sodium in blood volume. increased vasopressin retains water and constricts the blood vessels and it is made in the hypothalamus |
|
|
nephrogenic diabetes insipidus |
lack of vasopressin response in kidneys |
|
|
dipsogenic |
defective thirst regulatino |
|
|
gestational |
increased levels of vasopressinase during pregnancy |
|
|
diabetes mellitus in general |
based on hyperglycemia, problems with lipid metabolism, and excreting too much glucose in the urine. both have genetic and environmental factors |
|
|
type 1 DM |
lack of insulin with death of beta cells. diabetic ketosis leads to an earlier detection of the disease. (hyper production of ketone bodies). this is an autoimmune response - mostly genetic (HLA region) but could also be caused by an environmental factors. t cells destroy infected beta cells adn then eventually all of them. the remaining beta cells can compensate but only for so long. enviro factors include breast feeding, microbiome, vitamin d |
|
|
type 2 DM |
insulin resistance, muscle, fat, liver problems. most prevalent form. also has a genetic factor but not a single one - hard to pinpoint. everytime you secrete insulin you secrete the amyloid fibrils ! not good cause you're secreting more insulin. glucolipotoxicity |
|
|
diagnosis of DM |
normal insulin --> increased insulin and increased glucose --> no insulin and increased glucose . to test, you look at hemoglobin A1C in fasted state or you can do an oral glucose challenge |
|
|
type 2 DM physiological effects |
muscle: type 4 no longer works - liver: increased hepatic output - adipose: increased lipolysis --> increased FFA |
|
|
MODY |
diabetes in the young - monogenetic mutation. obesity not as frequent. diagnosis is much earlier |
|
|
gestational diabetes |
only while pregnant. not quite sure why this happens but it is also sort of because of the increase in hormones like progesterone |
|
|
insulin signalling |
1) insulin binds to receptor --> autophos and conformational change 2) irs 1binds to ptb domain which phos irs1 3) pi 3 binds to irrs1-p and phos other pi's 4) pi's bind to PKB and recruits it to membrane where it is phos and activates 5) activated pkb is released from membrane and promotes glut 4 uptake and glycogen synthesis (by PKB phos GSK3 which inactives GSK bucause GSK inhibit glycogen synthase) |
|
|
lipodystrophy |
complete lack of adipose tissue leads to severe insulin resistance and reqiures fat transplantation or leptin injections |
|
|
good fat |
non visceral fat (subcutaneous). good fat activates insulin sensitivity and increases leptin and adiponectin. decreases tag synthesis and increase beta ox and increases insulin |
|
|
bad fat |
visceeral fat. decreases insulin sensivity and activates proinflam like tnf alpha. visceral fat releases lots of fa |
|
|
adiponectin levels when obese |
decrease while leptin increases |
|
|
free fa effects |
in the liver: increases IR and increase gluconeogensis, inapropriate glucose release in the pancreas: increase insulin release acutely but chronically it causes beta cell death in muscle: insulin resistnace nad decreased glucose uptake |
|
|
insulin resistance and inflammation |
activated macrophages in skeletal muscle and liver release adipokines. macrophages explode in stressed cells --> release TNF alpha kuffner cells sense lipi accumulateion and lead to JNK and IKK beta proinflammatory cytokines --> NFkB |
|
|
er stress from obesity |
activate unfolded protein response. caused by too many TAG and FA breaking the membranes. leads to insulin resistance |
|
|
unfolded protein response |
1too much cholestrol and tag creates er stress which activates IRE1 which activates XBP1 causing enhancement of phospholpiid biosyntehsis which increase membrane biosynthesis. also activates jnk and ikk |
|
|
function of xbp1 |
reduces the free cholesterol to phospholipid ration - mitigating er stress |
|
|
PKR and er stress |
PKR phos eif2 - antagonizes SREBP activation to decrease cholestrol synthesis |
|
|
er stress and irs1 |
increased adipose promotes er stress. ire1 : jnk phos irs 1 which downregulates insulin signalling |
|
|
jnk |
phos irs 1 and 2 and inhibits signals through ap 1 |
|
|
ikk |
inhibits thru transcriptional events mediated by NFkB |
|
|
metabolic overload in the liver |
fa cannot be shuttled into mitochondria because of impaired oxidation due to increased malyonyl coa. too many fa tried to be mitagated by making tag but this causes er stress. also increased DAG which activates stress kinases --> IR |
|
|
metabolic overload in muscle |
accumulation of lc-coa's, dag, tag --> IR. glucose uptake goes down. lazy mitochondria cannot handle the increase in fa --> reactive oxidatives. triggers stress kinases like PKC --> phos irs1 --> IR |
|
|
diabetic ketoacidosis |
occurs in type 1 DM. low pH and hyperglycemia. ketone bodies are produced by the liver and a part of the normal fastng response. symptoms: deep sighing(a response of bicarbonate going to co2 as a buffer) and fruity breath due to protein denaturation. severe dehydration and poydipsia. results from absolute insulin def and hormones excess. treatment: hydrate with iv full and insulin |
|
|
hyperglycemic hypersomolar state |
in type 2 diabetes. similar to ketoacidosis. symptoms: dehydration, hyperosmolarity |
|
|
acute diabetic complications |
ketoacidosis and hyperglycemic hypersomolar state |
|
|
chronic diabetic complications |
nueropathy (eye disease) microvascular, macrovascular, and nonvascualar |
|
|
hyperglycemia |
extracellular --> nonenzymatic glycosolation of proteins --> AGES and HEMO A1C intracellular --> disturbances of the polyol pathway |
|
|
AGES what are |
end product of extracellular hyperglycemia / nonenzymatic glycosation. 1) reversible schiff base 2) irreversable crosslinked amadori products . |
|
|
AGES effects |
vessel wall stiffness, trapping protiens in the crosslinks including antibodies, resistance to degration, increased foam cells --> athersclorosis . increase vascular permeability and ECM production and cell proliferation |
|
|
polyol pathway:; why happen |
alternative handling pathway for glucose. creates sorbitol and fructose |
|
|
polyol pathway: machanism |
1) reduce by aldose reductase --> sorbitol 2) fructose and nadh |
|
|
results of the polyol pathway |
cannot get rid of sorbitol, increased cell osmolarity (cells burst), increased AGES, PCK activation, abnormal enzyme behavior due to insertion of glucose into the hexosamine pathways |
|
|
macrovascular complications |
arise from damages blood cells --> aterosclorosis |
|
|
types of microvascular complications |
retinopathy, neuropathy, nephropathy |
|
|
nephropathy |
causes cellular damages including thickening of the basement membremae, chronic renal failture. accelerated by hypertension. nephrons die and you leak out good things. leads to nephronic syndrome --> wasting ad protein loss --> infections, anemia, malnutrition, rickets, altered blood lipids. treatment: diet with moderate protein and low sodium |
|
|
neuropathy |
nuerons die and vascular occlusions. injuries go untreated. does not happen in brain. can lead to bacterial infections and GENGREENE and amputations |
|
|
retinopahty |
thickening of the baseent membrean and loss of pericytes which express Aldose reductase and control blood flow. this leads to hemorrages. maculopathy on macula reduce sight. capillary clsorue and angiogenseiss |
|
|
diabetic complications in pregnancy |
miscarriages, large babies, and babies with hypoglycemia |
|
|
what organ controls how much glucose tissues take up? |
liver |
|
|
what organ controls how much glucose tissues take up? |
liver |
|
|
glycogen |
can give muscles bursts of energy. has high water content. cannot be packed close. has 1,4 alpha bonds with 1,6 aloha branches. the braches act as fast response to low energy. |
|
|
what organ controls how much glucose tissues take up? |
liver |
|
|
glycogen |
can give muscles bursts of energy. has high water content. cannot be packed close. has 1,4 alpha bonds with 1,6 aloha branches. the braches act as fast response to low energy. |
|
|
how do u make glcogen |
glcogenin is the center residue. glucose 6 phos goes into UTP and g1p which adds together to make a UDP glucose. this autocatalyzes the first 5 residues but then glycogen synthase takes over. branching enzyme transfers 7/11 of glycogens into a branch. |
|
|
what organ controls how much glucose tissues take up? |
liver |
|
|
glycogen |
can give muscles bursts of energy. has high water content. cannot be packed close. has 1,4 alpha bonds with 1,6 aloha branches. the braches act as fast response to low energy. |
|
|
how do u make glcogen |
glcogenin is the center residue. glucose 6 phos goes into UTP and g1p which adds together to make a UDP glucose. this autocatalyzes the first 5 residues but then glycogen synthase takes over. branching enzyme transfers 7/11 of glycogens into a branch. |
|
|
what is an allosteric activator of glycogen synthase |
glucose 6 phos |
|
|
what organ controls how much glucose tissues take up? |
liver |
|
|
glycogen |
can give muscles bursts of energy. has high water content. cannot be packed close. has 1,4 alpha bonds with 1,6 aloha branches. the braches act as fast response to low energy. |
|
|
how do u make glcogen |
glcogenin is the center residue. glucose 6 phos goes into UTP and g1p which adds together to make a UDP glucose. this autocatalyzes the first 5 residues but then glycogen synthase takes over. branching enzyme transfers 7/11 of glycogens into a branch. |
|
|
what is an allosteric activator of glycogen synthase |
glucose 6 phos |
|
|
how is glycogen broken down into glucose? |
debrancher enzyme --> glycogen phosphorylase and vita B6 makes g6p --> muscle or liver to blood (via g6phosphatase) |
|
|
what organ controls how much glucose tissues take up? |
liver |
|
|
glycogen |
can give muscles bursts of energy. has high water content. cannot be packed close. has 1,4 alpha bonds with 1,6 aloha branches. the braches act as fast response to low energy. |
|
|
how do u make glcogen |
glcogenin is the center residue. glucose 6 phos goes into UTP and g1p which adds together to make a UDP glucose. this autocatalyzes the first 5 residues but then glycogen synthase takes over. branching enzyme transfers 7/11 of glycogens into a branch. |
|
|
what is an allosteric activator of glycogen synthase |
glucose 6 phos |
|
|
how is glycogen broken down into glucose? |
debrancher enzyme --> glycogen phosphorylase and vita B6 makes g6p --> muscle or liver to blood (via g6phosphatase) |
|
|
what is an inhibitor of glycoen synthase? |
ampk |
|
|
how does insulin and glucagon affect glycogen/g1p cycling? |
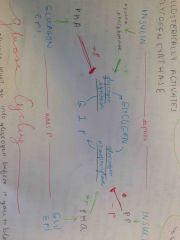
Back (Definition) |
|
|
what are the phosporases or phosphatases for glycogen synthase and glycogen phosphatase? |
pka and protein phosphatase |
|
|
what must glucose do before it can go into the blood stream? and why? |
be cycled through glycogen. because you never want to have no glycogen |
|
|
what do orecigenic signals do? and what is an example? |
increase appetite. agrp |
|
|
what must glucose do before it can go into the blood stream? and why? |
be cycled through glycogen. because you never want to have no glycogen |
|
|
what do orecigenic signals do? and what is an example? |
increase appetite. agrp |
|
|
what does an anorecigenic signal do? what is an example? |
stops feeding. pomc. |
|
|
what must glucose do before it can go into the blood stream? and why? |
be cycled through glycogen. because you never want to have no glycogen |
|
|
what do orecigenic signals do? and what is an example? |
increase appetite. agrp |
|
|
what does an anorecigenic signal do? what is an example? |
stops feeding. pomc. |
|
|
what does metformin do? |
stimulated ampk |
|
|
fox o1 and gluconeogenesis |
akt phos's fox o1 and it leaves the nucleus and it inhibits GNG during mild fasting or long term (sirt 1) de phos fox o1, fox o1 goes unto the nucleus and increases GNG through pepCK glucose is released into the blood and it activates sestrin which activates ampk which stimulates fa ox |
|
|
gluconeogenesis and CREB/CRTC2 |
when glucagon is high, it p's creb which is in dimer with crtc2 --> increases GNG/ when there is insulin, akt is active and p's SIK which p's CRTC2 which inhibits it |
|
|
von gierke's disease |
glycogen storage disease: growth failure, lacticidosis, hypoglycemia, g6pase |
|
|
pompe's disease |
glycogen storage disease: muscle weakeness, heart failure, glucosidase |
|
|
cori's/forbes |
glycogen storage disease: hypoglycemia, myopathy, growth retardation, debrancher |
|
|
andersen |
glycogen storage disease: cirrohsis, death by 5, branching |
|
|
mcardle's |
glycogen storage disease: exercise cramps, renal failure, muscle glycogen phosphorylase |
|
|
hers' |
glycogen storage disease: hypoglycemia, liver glycogen phosphorylase |
|
|
tarui's |
glycgen synthase disease: exercise cramps, weakness, growth retardation, phosphofructokinase |
|
|
brown adipose tissue characteristics |
multiocular, generate heat, endocrine function, limited, inverse correlation with outdoor temp, depensd on nutritional status, becomes less functional with ageb |
|
|
brown adipose functins |
inversely related with adipoisty and bmi. expresses UCP1. increases TAG clearance, generates heat via UCP1 via fa ox |
|
|
mechanism of UCP1 |
mitochondrial respiration, as yu make NAD+, o2--> h20 via ETC then ATP via ATP synthase. cells with BAT make HEAT insteaed of ATP |
|
|
is warm or cold better for BAT? |
cold |
|
|
UCP1 null mouse effects |
mouse lost weight because it is used to the fa methods of BAT but it is just doing it in ineffecient methods? |
|
|
regulatin of UCP1 |
1) sensed by sympathetic NS -->norepi --> beta adrenergic --> cAMP + PKA --> CRPP --> stimulates GNG and lipolysis. 2) thyroxine is taken up by BAT --> activates ppars |
|
|
type 1 nuclear receptors |
steriod receptors. in cytoplasm, binds, dimerizes and enters nucleus binds to DNA (HRE) --> activates transcription |
|
|
what nuclear receptor is for steriods? |
type 1 |
|
|
type 2 nuclear receptors |
for the other stuff like thyroid and -XRs. these are being sensed in larger quantities. already bound to DNA as heterodimers with HREs and RXRs. if there is no ligand, there is a corepressor atached. if there is a ligand, a co activator replaces it |
|
|
type 3 receptors |
binds to direct repeats |
|
|
type 4 receptors |
only bind a single DNA binding domain. binds to a single half site HRE |
|
|
where are glucocorticoids produced and what kind of nuclear receptor do they bind? |
adrenal gland on top of the kidney. type 1 (bcsteriod) |
|
|
effects of cortisol |
increased during fasting. stimulates fa metab, stimulates GNG and glucose release in the liver, inhibits uptake of glucose by muscle, increase breakdwn of muscle protien |
|
|
cushing's syndrome |
verprduction of cortisol. central obesity and insulin resistance. abnormal body shape and euphoria |
|
|
vitamin d pathway and where |
vita d -- 25 hydroxylase --> 25 OH D. happens in kidney. type 2 receptor. |
|
|
function of vitamin d |
calcium hmeostasis. release calcium from bone and reabsorb it in the kidney. prmotes calcium absorption in the intestine |
|
|
retinioc acid recptor |
type 2 recptor |
|
|
estrogen receptor |
type 1 effects glucse uptake by muscle, prevents visceral fat, increases central sensitivit of leptin, increases leptin receptors in adipose: prevents development of obesity and insulin resistnece |
|
|
thryoid hormone |
type 2. iodinated aa derivitives. synthesis and secretin stimulated by TSH . can decrease weight (increasing heart rate and body temperature), lwoer cholestrol. loss of TSH can cause cardiac problems, muscle wasting, bone fatigue, etc |
|
|
dietary fa regulation of gene trx |
by binding like with ppars or by indirectly changing like srebp |
|
|
ppars |
for fa and deritivies. three forms: alpha, gamma, delta. type 2. no fa --> n transcription. |
|
|
ppar alpah |
activated by unsta fa and fibrates. increases mito and peroxisomal breakdown of FA. increases lipid oxidation, ketogenesis and glucose sparing (to help fight starvation). increases HDL. found in liver, heart, kidneys, BAT |
|
|
ppar gamma |
activated by fa, prostoglandinds and txds (inflam). stimulates differentiation of adipocytes and fa uptake and storage. changes adipokine expression. (increases adiponectin but decreases leptin) increase adipose size and more adipose in the right places. |
|
|
ppar delta/beta |
activated by long chain fa. increases muscle's fa ox and switch from type 2 to type 1 muscle fibers. in adipose, skin, brain, everywhere. |
|
|
endocrine control of feeding is regulated where? |
arcuate nucleaus in the brainstem/hypothalamus |
|
|
agrp/npy |
orexigenic - make u hungry ! they increase food uptake by changing behavior |
|
|
npy |
binds to a gcpr. stimmulated by fasting. you will not immediately starve without it because of backup mechanisms |
|
|
agrp |
in skin color and fur (agouti) binds to melocortin receptrs and coexpressed by npy. long term --> obesity. |
|
|
orexins |
two peptides linked to feeding, sleep and activity. delays onset of satiety. if you have a low level you stop eating earlier. |
|
|
disruption of orexins |
narcolepsy |
|
|
reward system in response to feeding |
rewarding nature of food. stimulates feedings. incolces opiods, endocann, dpaminergic and serotonin. |
|
|
rimbonabant |
antagonizes reward system - antiobesity drug |
|
|
pomc/cartt |
anoerectic signals telling you are full and should not eat. |
|
|
cart |
wide spread in the CNS. food intake, bdy weight, reward ,endocrine functions. in arc expression decreased with fasting. also reduces refeeding response. blocks npy singal. mutatins leads to obesity |
|
|
pomc |
expressed in tissues. in arc, msh alpha goes to the melocortin receptors/pathways (MC4) |
|
|
MC4/melocortin receptor |
how does it work with both of them? agonizing --> increases cAMP. agrp disrupts the active form and there is no activation. |
|
|
effects of losing mc4 |
obestity |
|
|
perihperal signals coming from the gi tract in response to feeding |
gherlin and GLPg |
|
|
gherlin |
gherlin receptors are on agrp/npy. released by empty stomach. also increased by lasck of sleep. it is released right before you would normally eat and supressed at night. regulated by circadian rhyhm |
|
|
GLP |
glucagon like peptide. from intesetine - released after intestine senses different nutrients. prepares body for carb rich meal. stimulates insulni release and inhibits appetite and food intake. |
|
|
signals frm pancreas in resposne to feeding |
insulin, pp, somatostatin |
|
|
somatostatin |
is released by beta cells in response to high glucose and alanin . inhibits gi secretions |
|
|
pp |
released by islets after eating. reduces food intake and slows absrorption |
|
|
adipokines in response to feeding |
for long term regulation. leptin, ampk |
|
|
leptin |
exclusively produced by adipose and sensed by hypothalamus. more leptin suppresses fedeings and down regulates npy and mcs, orexins, and sgrp. upregulates the other ones. |
|
|
what activaets leptin |
mtor |
|
|
ampk in response to feeding |
fuel sensor. integrates all the signals. |
|
|
characteristics of alzheimemrs |
amyloid fibesr and nueronal tangles (microtubulues - phos tau --> destabilize and accumulates inside nueron ) |
|
|
molecular mechanisms of AD |
1) systemic defeciency --> impairs learning prcess 2) in brain, insulin cpmpeptes with AB for insulin degrading enzymes --> amyloid plaques build up 3) IR --> hyperphos tau --> nueronal tangels |
|
|
amyloid plaques |
can build up in the brain during AD. cause a positive feedback loop that enhances phos of tau and nueronal tangels. |
|
|
apo e2 |
duoble cys. can make CVD workse because you cannot clear the VLDL particles --> inflam --> heart disease |
|
|
apo e4 |
is assciated with AD. because the brain moves lipids around and astrocytes are improtant for this. astrocytes need nutrients and apo e4 are not giving it to the,/ . |
|
|
which apo e makes AD wrse? |
e4 |
|
|
how to treat AD |
exercise? lowering choelstrol, ppar agonists, insulin and glp receptor agonists can reverse hte beta amlyoid fibers and insulin resistance. intranasal insulin --> straigh tot the dome. |
|
|
first line of defense against diabetes |
exercise and good diet. |
|
|
lifestyle management for diabetes |
dietary habits, physcial activity, behavior modification |
|
|
mediterranean diet |
high fiber low fat |
|
|
physical activity |
moderate - best if yu do with diet |
|
|
insulin replacement therapy |
fast activing - right before meal. slow release - befre bed. gd for type 1 diabetes. con: if you inject too much you can becoe hypoglycemic. and u must inject a lot so a lot of people do not keep up with it. |
|
|
sulphonylureas |
diabetes treatment - for type 2 - for stimulating endogenous insulin production. inhibits k+ channels causing depolarization of ca++ channels leading to insulin release. side effects: could cause weight gain + hypoglycemia. it could also exhaust beta cells |
|
|
stimulate glp1 signals |
diabetes treamtent. glp is produced by the small intestine. makes you feel full and increase the amt of insulin released. a synthetic version is degraded quickly . delays gastric emptying |
|
|
dipeptydl peptidase 4 inhibitors |
stablilize endogenusly released glp1 so it is not degraded as quickly . you wouldn't have to inject as much insulin. |
|
|
metformin |
stimulates ampk . increase glucse uptake enchances fa ox and decreased glucse being relae. insulin sensitizer. |
|
|
fibrates |
ppar alpha agonists. diabetes treatment. liver can break down more fa by making more mitochondria. increase beneficial HDL and decrease TAG |
|
|
glutazones |
type 2 receptor - ppar gamma agonist. increases size of adipocytes --> increase good fat. caused heart attacks. used for diabetes treatment. |
|
|
ppar delta agonists |
diabetes treatment. highly expressed in muscle. increase HDL decrease TAG and chlestrol |
|
|
statins |
ultimately lowers LDL. huge drug |
|
|
inhibit reabsorption o f glucse in the kidney |
diabetes treamtment. glucose is reabsorbed thru SGLT2. can cause bladder infections. |
|
|
fen-phen |
diabtets treatment. increases seratonin levels whichahs anorertic effects. causes heart problems. |
|
|
alpha glucosidase |
inhibits digestion of glucose and not proteins |
|
|
orlistat |
binds to the active site of pancreatic lipase. prevents genertaion of FA --> gives to colon. nasty po. |
|
|
ezetimibe |
blocks dietary cholestrol by blocking trasnprter |
|
|
sibutramine |
causes satitety by increasing serotnin thru inhibition of reuptake |
|
|
DNMT1 |
makes sure that a methyl group stays on one of the DNA strands durign replicatin. |
|
|
methylation in epigenetics |
germ cells have some methylated patches. there is a global wave of demethylaation and then ne that is sex specific this process is driven by development and differentiation |
|
|
exceptions in demethylation (epigenetics) |
patterns for sex specificity are not erased |
|
|
what effect does methylation have on genes |
supresses them |
|
|
what is required for methyaltion |
folate / folic acid - acts as a methyl donor |
|
|
agouti mice deomstrate what principle? |
epigenetics - range of colors means that there is differences in methlyation and transposns |
|
|
what must glucose do before it can go into the blood stream? and why? |
be cycled through glycogen. because you never want to have no glycogen |
|
|
what do orecigenic signals do? and what is an example? |
increase appetite. agrp |
|
|
what does an anorecigenic signal do? what is an example? |
stops feeding. pomc. |
|
|
what does metformin do? |
stimulated ampk |
|
|
fox o? |
not quite sure what it does but it is involved in feeding. liver: it regulates glucose and lipid metab --> prevents fa syn beta cells: regulates proliferation, differentiation and stress resistance arn - not sure! |