Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
77 Cards in this Set
- Front
- Back
|
What are the 2 main types of kidney/bladder tumors?
|
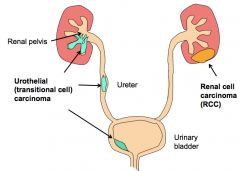
*RCC accounts for 85%.
*Urothelial is the second most common. 10-15%. *Renal cell carcinoma (RCC) is the most common primary malignant neoplasm of the kidney in adults (85% of renal cancers), but can occur in children. *Urothelial carcinomas of the renal pelvis are the 2nd most common (5-10% of renal cancers). |
|
|
What are the 4 main types of RCC?
|
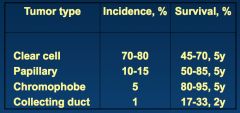
|
|
|
What is survival like in the different types of RCC?
|
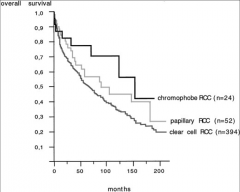
|
|
|
Describe the Clinical Presentation of RCC:
Where may there be mets? |
*Classic Triad (but only presents in 10% of cases):
1) Hematuria. 2) Costovertebral pain. 3) Palpable mass. *Often asymptomatic growth. *In 25% of RCC, there are metastases at presentation: -Lung (50%). -Bone (33%). *Patients can also present with Paraneoplastic syndromes: -Polycythemia -Hypercalcemia -Hypertension -Hepatic dysfunction -Feminization -Masculinization -Cushing syndrome |
|
|
What cells do RCCs arise from? What part of the kidneys do they affect most?
At what age do they usually strike? What gender? What are the risk factors? |
*Derives from renal tubular epithelium.
*Most commonly affects the poles. *Most commonly 6th and 7th decades. *M:F=2:1. *Risk factors: -Smoking. -Hypertension. -Obesity. -Heavy metals (cadmium). -Acquired polycystic kidney disease as a result of chronic dialysis (30x risk). -Tuberous sclerosis. |
|
|
How do you distinguish between the different types of RCCs?
|
*There are distinct histological subtypes with characteristic gross and microscopic appearance.
*Distinct histogenesis. *Unique cytogenetic abnormalities (at the level of the chromosome). *Chromosomal deletions, amplifications, translocations and/ or genetic abnormalities (at the level of the gene). *There can be loss of function mutations in tumor suppressor genes (VHL, BHD). *There can be activation mutations in oncogenes (MET). |
|
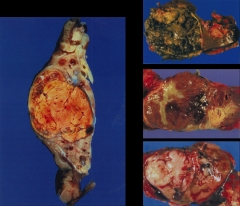
|
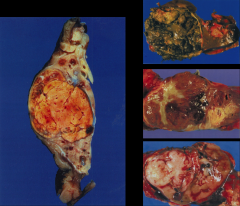
*Different types of RCCs.
L: CCC. TR: Middle: |
|
|
Looking at the nephron, identify what parts give rise to different kinds of tumors:
|
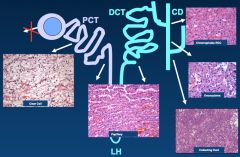
*Most common is CCC; arises from PCT.
*Chromophobe and Oncocytoma come from the intercalated cells of the CD. *No one knows where papillary comes from; shares traits of PCT and DCT. |
|
|
Discuss the Clear cell RCC:
|
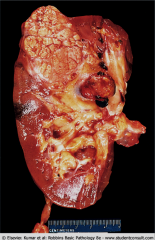
*Most common type of RCC; ~70-80%.
*Arises from proximal tubular epithelium. *Gross: -Usually solitary, ranging from 3-15 cm. -Yellow-orange (lipid). -Hemorrhages. -Can have cystic change. -Well circumscribed. -They show invasion into the renal vein. |
|
|
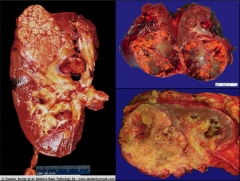
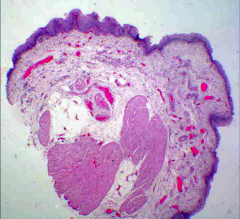
*Gross specimens of CC RCC.
*Left pic shows tumor in the renal vein. |
|
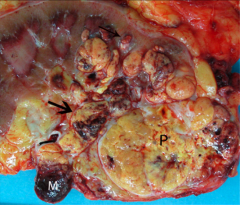
|
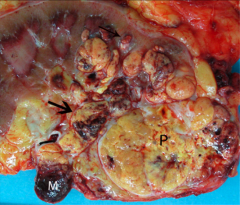
*Renal Vein Invasion in CC RCC. This can progress into the IVC and into the heart!
|
|
|
How do we stage RCCs? Why is this important?
|
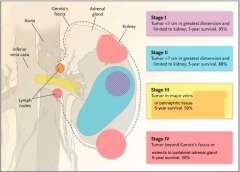
*Renal vein involvement = stage 3.
*Staging correlates with survival. *Tumor size, lymph node involvement, and mets are used to stage. |
|
|
What are the µscopic traits of Clear Cell RCC?
|
*Clear cytoplasm (due to high lipid, glycogen), may have granular cytoplasm.
*Distinct cell membrane. *Growth patterns: 1) Solid 2) Trabecular (cordlike) 3) Tubular *Stroma usually scant. *Highly vascularized stroma. |
|
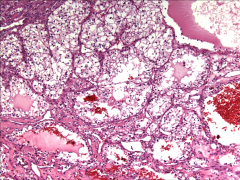
|
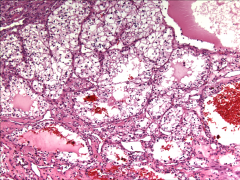
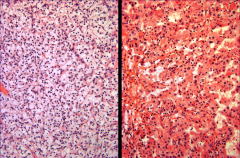
*Clear Cell RCC.
*Note clear cytoplasm and rich vasculature. |
|
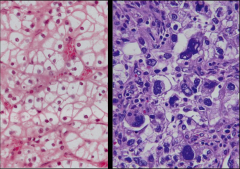
|
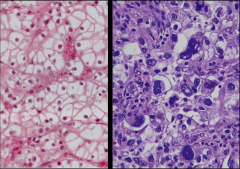
*Clear Cell RCC.
*L: Low grade. Better differentiated. Nuclei aren't so large. *R: High grade. Poorly differentiated. Nuclei are huge. |
|
|
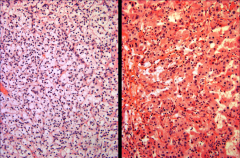
L: Clear cytoplasm in CC RCC.
R: Granular cytoplasm in CC RCC. *Cytoplasm doesn't HAVE to be clear! |
|
|
What are the different patterns we see in sporadic vs. inherited kidney cancers?
-Age of onset? -Frequency of each? -Pattern of kidney involvement? -Type of mutation? |
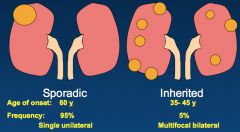
|
|
|
Discuss VHL disease:
-Inheritance? -Where all do we see tumors? |
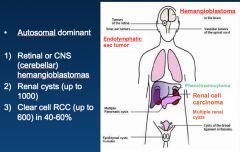
1) Hemangioblastoma
2) Pheochromocytoma 3) RCCs-- ALL CLEAR CELL! *Each patient is different, and may not have all the involvement shown on this slide (VHL gene can have different kinds of mutations). |
|
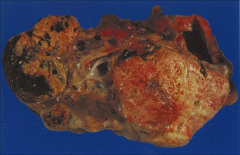
|
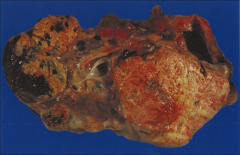
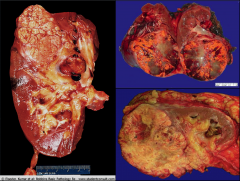
*Gross kidney in VHL. Shows multiple cysts and multiple tumors. You can see the yellow color that's characteristic of CC RCC.
|
|
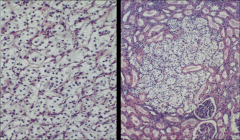
|
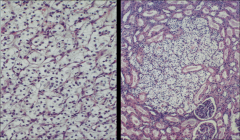
*CC RCC in VHL disease.
*Note small size of the tumor on the right; this tumor is likely 1-2mm in diameter. Remember that in VHL disease the patient can have HUNDREDS of these. |
|
|
How is the mutation for CC RCC acquired in VHL (like from the perspective of "hits")?
|
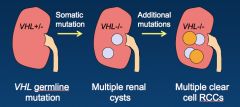
*Short arm of chromosome 3.
*There's a GERMline, universal mutation. *There's a loss of function mutation in the VHL tumor suppressor gene in the kidneys--> bilateral tumors. |
|
|
How is the mutation for CC RCC acquired in sporadic cases (like from the perspective of "hits")?
|
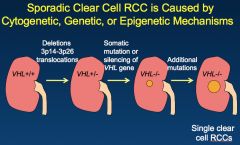
*Kind of similar; still involves short arm of chromosome 3, and 80% of cases still involve the VHL gene.
*NO GERMLINE MUTATION. *First event: deletion of 3p. *Second event: somatic mutation or epigenetic silencing; loss of VHL function. *Tumors are SINGLE AND UNILATERAL. |
|
|
What is the function of VHL gene? What goes wrong when it gets mutated?
|
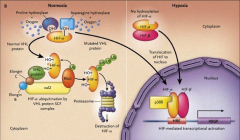
*Abnormal VHL Protein Causes Accumulation of Hypoxia Inducible Factor-a.
*VHL PRO is normally part of the ubiquitin system. When the ubiquitin system doesn't work, HIF accumulates. |
|
|
What's the significance of HIF accumulation in VHL mutations?
|
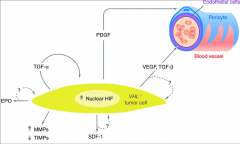
*HIF Stimulates Tumor Cell Growth and Angiogenesis--> VEGF.
*There's a pathway of steps involved (PDGF, TGF, etc.). |
|
|
Discuss Papillary RCC:
How common? What forms are there? Where are the tumors? Who gets this? |
*15% of RCCs.
*Familial and sporadic forms. *Frequently multifocal and bilateral. *Share phenotype of distal and proximal tubules. *The most common type of RCC in patients who develop dialysis-associated cystic disease. |
|
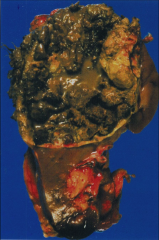
|
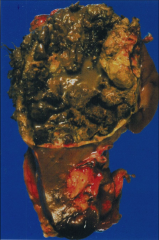
*Papillary RCC.
*Good example of a gross specimen: -Well circumscribed -Encapsulated -Hemorrhagic -Cystic *If there's any yellow color present, it's from macrophages. |
|
|
What are the µscopic features of Papillary RCC?
|
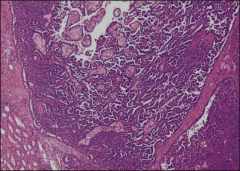
*Papillary formations.
*Cuboidal to low columnar cells. *Foamy macrophages in papillary cores. *Psammoma bodies (concentric calcifications) may present. *Highly vascularized stroma. |
|
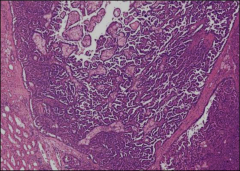
|
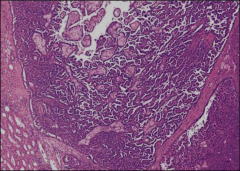
*Papillary RCC. On the left you see normal kidney tubules. Tumor is well-cirumscribed and encapsulated.
*Note papillae (with white space between). *Pale cells in the middle are foamy macrophages. |
|
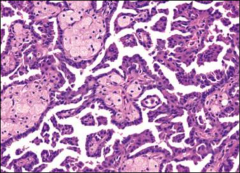
|
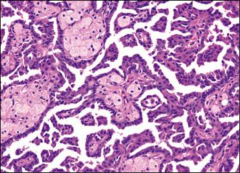
*Papillary RCC, high power.
*Foamy macrophages are the light objects located within the papilla. |
|
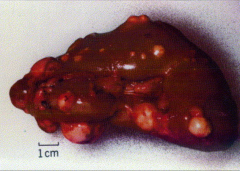
|
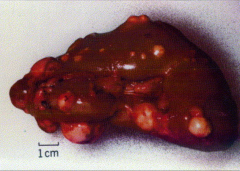
*Hereditary Papillary Renal Cell Carcinoma (HPRCC).
*Bilateral. Multiple tumors. |
|
|
Discuss HPRCC:
|
*Autosomal dominant.
*Late onset. *Germline ACTIVATING mutations in Met protooncogene (7q31). *Tumors: selective duplication of mutant allele in chromosome 7. *Multifocal (may be >100 tumors). *No extrarenal manifestations. |
|
|
How are mutations acquired in HPRCC?
|
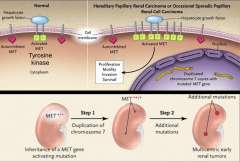
*Becomes activated without hepatocyte growth factor, which is normally required to activate it.
*Starts with a GERMline mutation (trisomy 7), followed by a gain of function of MET. |
|
|
What are the cytogenetic differences between the sporadic and inherited forms of HPRCC?
|
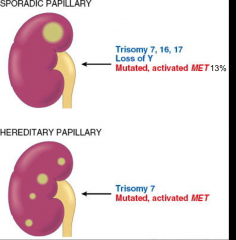
*Sporadic is rare, but look at all the trisomies plus loss of Y.
|
|
|
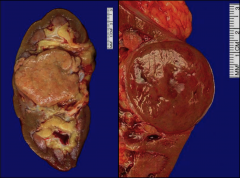
Discuss Chromophobe RCC:
Where does it originate? Gross traits? |
*5% of RCC
*Originate from intercallated cells of collecting ducts. *Excellent prognosis. *Gross: -Well circumscribed. -Tan/ Mahogany. -Rarely, hemorrhage or necrosis. |
|
|
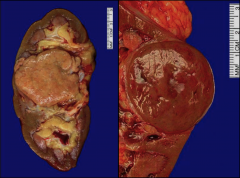
*Chromophobe RCC. Well circumscribed.
|
|
|
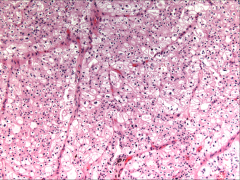
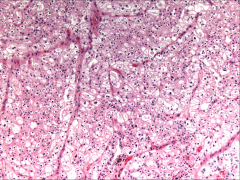
*Chromophobe RCC. Cells are LARGE, and they contain some eosinophilic cytoplasm. They line up along capillaries.
|
|
|
Discuss the µscopic characteristics of Chromophobe RCC:
|
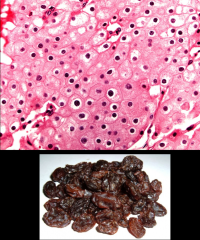
*Solid sheets.
*Concentration of large cells around blood vessels. *Prominent cell membranes. *Pale eosinophilic cytoplasm. *Irregular, wrinkled, rasinoid nuclei. *Perinuclear halo. |
|
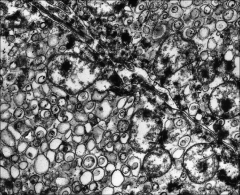
Discuss the characteristics of Chromophobe RCC on EM:
|
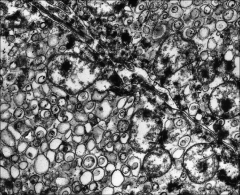
*Cytoplasm is filled with abundant microvesicles (maybe from degradation of mitochondria).
*The few mitochondria are localized near to the cell membrane. |
|
|
Discuss the ∆ b/t the inherited and sporadic forms of chromophobe RCC:
|

*Inherited is rare. Associated with BHD mutation.
*Sporadic RARELY involves BHD mutation; usually involves chromosomal abnormalities. Don't have to remember which ones are involved. |
|
|
Discuss oncocytoma:
What might it get confused with? |
*5% of renal neoplasms. Rare.
*Originates from intercalated cells of collecting ducts (just like chromophobe RCC). *BENIGN tumor with excellent prognosis. *Main differential diagnosis with chromophobe RCC. |
|
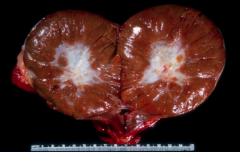
|
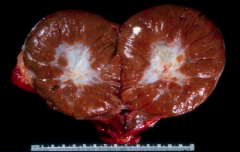
*Oncocytoma.
*Mahogany brown with a central scar. |
|
|
What are the µscopic traits of oncocytoma?
|
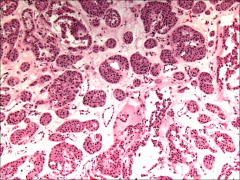
*Nesting pattern
*Round to polygonal cells *Eosinophilic cytoplasm *Small round nuclei *Large nucleoli *Mitotic figures only rarely identified |
|
|
|
|
|
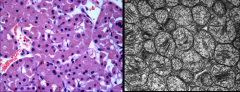
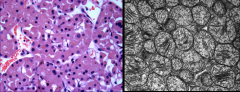
*Oncocytoma.
L: Shows Granular eosinophilic cytoplasm. Granularity is due to huge amounts of mitochondria. R: Mitochondria on EM. |
|
|
Describe the significance of Birt-Hogg-Dube Syndrome :
What organs are involved? |
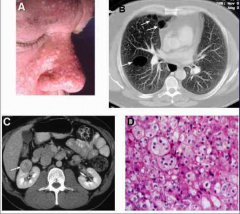
*Caused by Mutations in BHD Gene.
*Benign skin tumors: -Fibrofolliculoma -Trichodiscoma -Acrochordon *Pulmonary cysts. *Renal cell tumors (Chromophobe RCC, oncocytoma, hybrid tumors with features of both.) *BHD gene is on chromosome 17l produces the protein folliculin. |
|
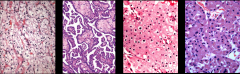
ID these tumors and their gene association:
|
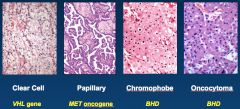
|
|
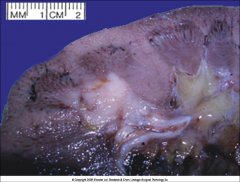
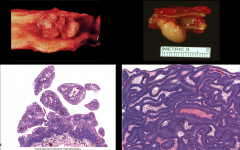
ID and discuss this tumor:
|
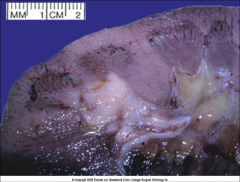
*Collecting Duct (Bellini Duct) RCC.
*RARE, 1% of all RCC. *Arise from collecting duct cells in medulla *Gray-white and firm *Poor prognosis |
|
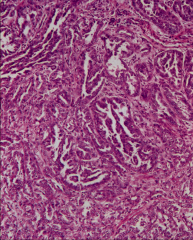
ID this tumor and discuss the features that are present:
|
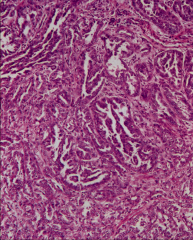
*Tubulopapillary architecture.
*Single cuboidal layer with hobnails. *Marked nuclear pleomorphism. *Desmoplastic and inflamed stroma (distinguishes this tumor). *Dysplasia of adjacent tubules. *HE SAYS THIS WON'T BE ON THE EXAM* |
|
|
Discuss Renal medullary carcinoma:
Who gets them? What's the gene association? How does the tumor behave? |
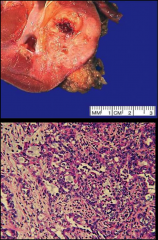
Young African American males (mean age 22 y).
*Sickle cell trait (HbAS) in 85% (may be a board question). *Located in medulla. *Originates in collecting ducts. *Highly aggressive; patients die in a couple of months. *May be a subtype of collecting duct carcinoma. |
|
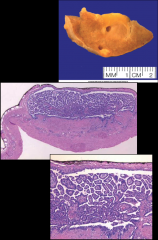
ID and discuss this tumor:
|
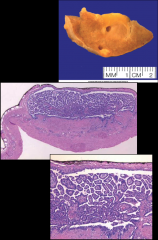
*Renal Papillary Adenoma.
*Benign. *Small (<0.5 cm...if you saw this same tumor and it was >0.5cm, this would by definition be a papillary renal cell carcinoma). *Histologically indistinguishable from papillary RCC. *NOTE: Clear cell, chromophobe, collecting duct-- these are never called adenomas even if they are small. |
|
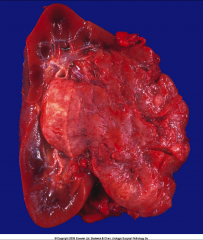
ID and discuss this tumor:
*Who gets these? |
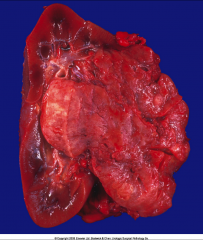
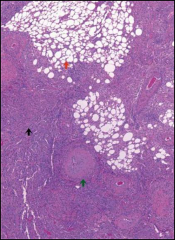
*Angiomyolipoma (composed of vessels, muscle, and fat).
*Benign. *Sporadic or syndromic. *Present in 25-50% of patients with tuberous sclerosis (seizures, mental retardation, autism, and tumors in many organ systems, including the brain, retina, heart, kidney, and skin). *Susceptible to spontaneous hemorrhage. |
|
|
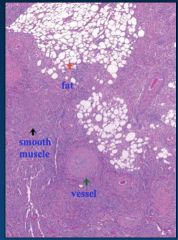
Angiomyolipoma
|
|
|
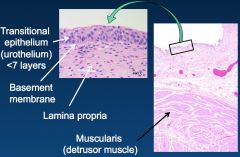
*This is NORMAL!
|
|
|
Discuss Tumors of the Urinary Bladder:
|
*Bladder is the most common site of urinary tract tumors.
*Older patients (median age 65 y). *M>F (3:1) *Almost always sporadic (familial is extremely rare). *90% are urothelial carcinomas (SEE ROBBINS 976). *Often multifocal. |
|
|
What are the Risk Factors for bladder cancer? 5
|
*Cigarette smoking (3-7 fold increased risk).
*Occcupational carcinogens (arylamines). *Schistosoma hematobium (Egypt, Sudan, other African countries) --> SCC. *Drugs (cyclophosphamide and analgesics). *Radiation therapy. |
|
|
How does bladder CA present?
|
*Painless hematuria is the dominant clinical presentation.
*Lesions that invade the ureteral or urethral orifices cause urinary tract obstruction. |
|
|
What are the 2 shapes of urothelial lesions?
What's the difference between them clinically? |
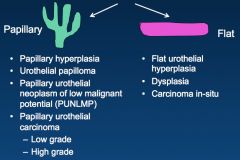
*Papillary urethelial carcinomas are never called carcinoma "in situ." We call them "non-invasive papillary carcinoma."
|
|
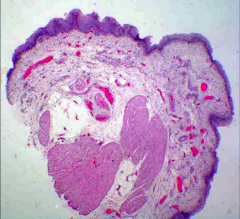
ID and discuss this entity:
|
*This is Papillary Hyperplasia.
*Urothelium is thicker on the left (>7 layers), but no need to count the number of cell layers. *There are undulating folds. *There are no fibrovascular cores. |
|
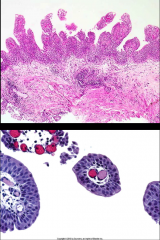
ID and discuss this lesion:
|
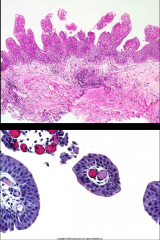
*Urothelial Papilloma.
*<1% of bladder tumors *Younger patients *Usually single lesions *Discrete papillary growth *Central fibrovascular cores *No atypia *Rare recurrences or progression. Benign. |
|

What's the difference?
|
*These are the 2 types of urothelial papilloma.
*Left: Exophytic (growing up within the lumen of the ureter). *Right: Endophytic (inverted papilloma; growing down). |
|
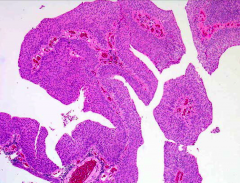
*Discuss this entity:
|
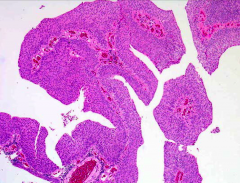
*PUNLMP.
*Somewhere in b/t malignant and benign. *Thickened urothelium (> 7 layers). *Orderly arrangement of cells within papillae. *Minimal architectural abnormalities. *Minimal nuclear atypia. *Mitotic figures rare. *May recur or rarely progress to high- grade tumors. |
|
|
Low-Grade Papillary Urothelial Carcinoma
|
|
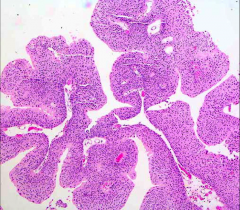
ID this and describe the features that you are seeing:
|
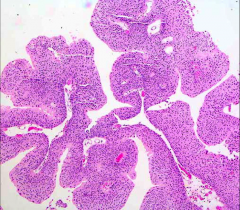
*Low-Grade Papillary Urothelial Carcinoma.
*Thickened layers (>7). *Orderly appearance. *Minimal but definitive cytologic atypia. You see SOME mitoses and some signs of malignancy. *OFTEN recur. *USUALLY non-invasive (90%). |
|
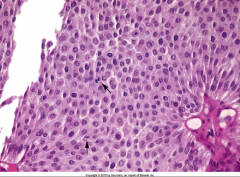
ID this and describe the features that you are seeing:
|
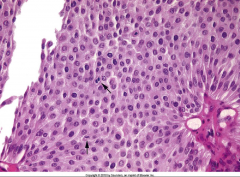
*High power view of Low-Grade Papillary Urothelial Carcinoma.
*Cells maintain polarity. *Scattered hyperchromatic nuclei. *Infrequent mitotic figures. *Mild variation in nuclear size and shape. |
|
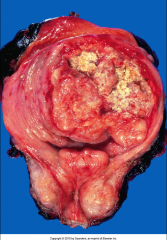
ID and discuss this tumor:
|
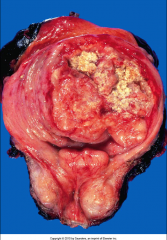
*High-Grade Papillary Urothelial Carcinoma.
-High incidence of invasion into muscularis (detrusor muscle). -Higher risk of progression than low-grade lesions. -Significant metastatic potential. |
|
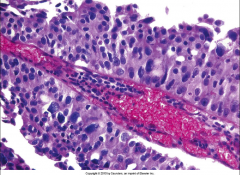
ID this and describe the features that you are seeing:
|
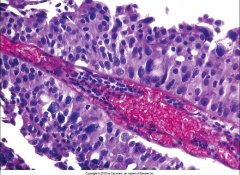
*High-Grade Papillary Urothelial Carcinoma.
*Architectural disarray and loss of polarity. *Nuclear atypia, some huge nuclei. *Frequent mitotic figures. |
|
|
Papilloma:
PUNLMP: LGPUC: HGPUC: 1) What's the recurrence of each? 2) How likely are each to invade? 3) How likely are each to kill you? |
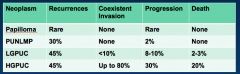
|
|
|
How do we stage bladder cancers?
|
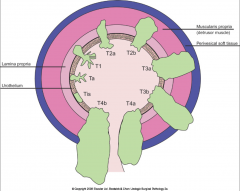
*Based on DEPTH of INVASION, not on tumor size.
*Papillary is called Ta, not Tis. *Note the layers involved with different stages. |
|
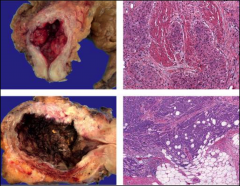
ID and give approximate staging:
|
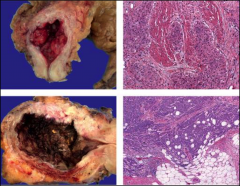
Top: Has invaded muscularis-- T2.
Bottom: Has invaded fat--T3. |
|
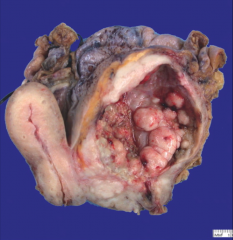
ID this and gives its approximate staging:
|
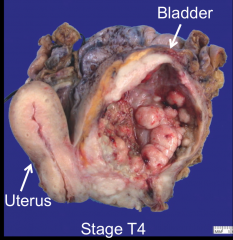
*Invasive Urothelial Carcinoma. Invading the uterus.
|
|
|
What is the survival of Urothelial Carcinomas, based on staging?
|
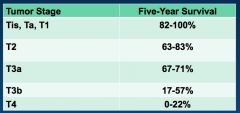
*The extent of spread (staging) at the time of initial diagnosis is the most important prognostic factor.
|
|
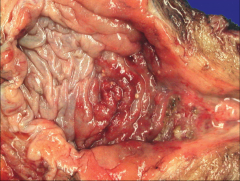
ID and discuss this entity:
|
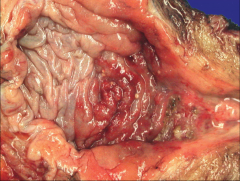
*Urothelial Carcinoma In-Situ.
*FLAT; no gross mass. *You see mucosal reddening, granularity, or thickening. *Commonly these tumors are multifocal. *May involve most of the bladder surface. |
|
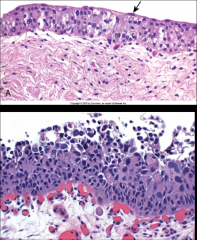
ID and discuss features:
|
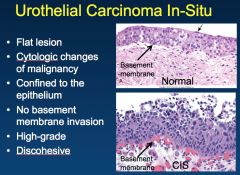
|
|
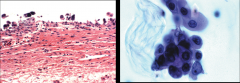
ID and discuss:
|
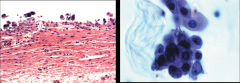
*Urothelial Carcinoma In-Situ.
*Left shows denuding CIS. *Right shows shedding of malignant cells into urine. *THESE ARE AGGRESSIVE TUMORS; 50% OF THEM WILL PROGRESS. *Urine samples can be used to track the tumor growth; it's difficult to do this in low grade ones. |
|
|
What's the genetic association of Urothelial Carcinoma In-Situ?
|
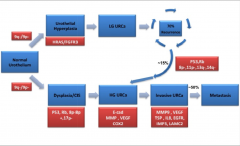
*Deletion of chromosome 9!!!! p16 gene is lost.
*Note difference in progression with low grade and high grade tumors. *Note that 15% of low grades INVADE; this is due to an additional mutation of p53 and Rb!! Low grade becomes high grade this way. *SEE ROBBINS 980. |
|
What are these variants of? Discuss.
|
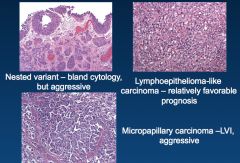
*Variants of urothelial carcinoma.
*These aren't terribly important for the test. |
|
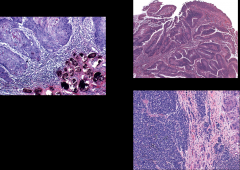
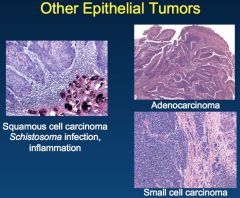
What are these? One of them has a connection to micro/ID.
|
|