![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
43 Cards in this Set
- Front
- Back
|
Chromosomenaberrationen ・Definition (3) |
- Abweichung von der normalen Chromosomenzahl (Aneuploidie) oder Chromosomenstruktur (z.B. Deletion, Translokation) - häufigste Grund für spontane Aborte |
|
|
Chromosomenaberrationen ・Übersicht (3) |
- Autosomale Chromosomenaberrationen → Trisomie 13 (Pätau-Syndrom) → Trisomie 18 (Edwards-Syndrom) → Trisomie 21 (Down-Syndrom)
- Gonosomale Chromosomenaberrationen → Klinefelter-Syndrom → Monosomie 45 (X0): Ullrich-Turner-Syndrom
- Weibliche Gonadendysgenesien ohne numerische Chromosomenaberrationen → Swyer-Syndrom → Reine Gonadendysgenesie 46,XX |
|
|
Triploidie ・Definition (1) |
- Vorliegen drei vollständiger Chromosomensätze |
|
|
Trisomie ・Definition (1) |
- Dreifaches (statt zweifaches) Vorliegen eines Chromosoms oder eines Chromosomenanteils |
|
|
Autosomale Chromosomenaberrationen ・Übersicht (3)
|
- Trisomie 13 (Pätau-Syndrom) → Ca. 1:10 000 (Korrelation mit dem Alter der Mutter) - Trisomie 18 (Edwards-Syndrom) → Häufigkeit: Ca. 1:6000 (Korrelation mit dem Alter der Mutter) - Trisomie 21 (Down-Syndrom) → 1:1500 - 6 (Korrelation mit dem Alter der Mutter) |
|
|
Pätau-Syndrom (Trisomie 13) ・Karyotyp (2) |
- ♀: 47, XX+13 - ♂: 47, XY+13 |
|
|
Pätau-Syndrom (Trisomie 13) ・Klinik (6) |
- Typisch ist die Trias aus → Mikrozephalie, Mikrophtalmie, evtl. Kolobome → Lippen-Kiefer-Gaumenspalte → Ulnare Polydaktylie, meist Hexadaktylie
- Weitere mögliche Symptome → Eventuell fehlgebildete Ohren, kleines Kinn, Vierfingerfurche, usw. → Kapilläre Hämangiome → Fehlbildungen innerer Organe: Angeborene Herzfehler (vor allem VSD, PDA), Nieren- und Harnleiterfehlbildungen |
|
|
Pätau-Syndrom (Trisomie 13) ・Prognose (1) |
- Nur 5% der Patienten werden älter als 6 Monate |
|
|
Edwards-Syndrom (Trisomie 18) ・Karyotyp (2) |
- ♀: 47, XX+18 - ♂: 47, XY+18 |
|
|
Edwards-Syndrom (Trisomie 18) ・Klinik (6) (Bild) |
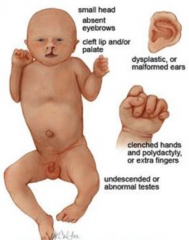
- Typische Handstellung mit Überlagerung von Mittel- und Ringfinger durch den Zeigefinger und kleinen Finger - "Faunenohren": Tief ansetzende, nach oben ausgezogene Ohrmuschel - Langer, schmaler Schädel - Mikrognathie, Lippen-Kiefer-Gaumenspalte, Hoher Gaumen, breite Nasenwurzel - Wiegenkufen-Füße - Fehlbildungen innerer Organe: Angeborene Herzfehler (vor allem VSD, ASD, Fallot-Tetralogie), Zwerchfellhernien, Nieren- und Harnleiterfehlbildungen |
|
|
Edwards-Syndrom (Trisomie 18) ・Prognose (1) |
- Nur 5% der Patienten werden älter als 12 Monate |
|
|
Trisomie 21 (Down-Syndrom) ・Einteilung (3) |
- Freie Trisomie 21 (ca. 95%) → ♀:47,XX,+21, ♂:47,XY,+21
- Translokationstrisomie 21 (ca. 4%) → ♀:45,XX,t(21;14), ♂:45,XY,t(21;14)
- Sonderform (ca. 1%) → Mosaik-Trisomie: ♀: 46,XX/47,XX,+21 bzw. ♂:46,XY/47,XY,+21 |
|
|
Freie Trisomie 21 ・Ätiologie (4) (Bild) |
- keine Erbkrankheit im engeren Sinne - In ca. 70% der Fälle bei der ersten, in ca. 20% bei der zweiten meiotischen Teilung der Eizelle durch Non-Disjunction - Kann durch väterliche Non-Disjunction in der Spermatogenese entstehen (in ca. 5% der Fälle) - Risiko für eine freie Trisomie 21 steigt mit dem Alter der Mutter an |
|
|
Translokationstrisomie 21 ・Ätiologie (3) (Bild) |
- Die Erbinformationen des Chromosoms Nummer 21 ist dreifach vorhanden, jedoch einmal an ein anderes Chromosom (meist Chromosom 14) angelagert - Nicht vom Alter der Mutter abhängig, familiär gehäuft (kann aber auch als Neumutation auftreten) - 50% der Fälle durch eine Vererbung einer balancierten Translokation (Robertson-Translokation) von einem Elternteil (meist der Mutter → Nicht an Trisomie erkrankt, unauffälliger Phänotyp) und in ca. 50% der Fälle durch Neumutation während der Meiose |
|
|
Trisomie 21 (Down-Syndrom) ・Epidemiologie (Tabelle 4) |
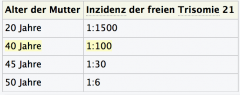
|
|
|
Trisomie 21 (Down-Syndrom) ・Klinik (5) |
- Merkmale an Gesicht und Kopf (kraniofaziale Dysmorphie) - Merkmale der Extremitäten, des Weichteilgewebes und des Skeletts - Organfehlbildungen - Körpergröße und Entwicklung
- Die Ätiologie der Trisomie 21 hat - mit Ausnahme der Mosaik-Trisomie - keinen Einfluss auf die Systematik der klinischen Symptome und den Krankheitsverlauf! |
|
|
Trisomie 21 (Down-Syndrom) ・Merkmale an Gesicht und Kopf (kraniofaziale Dysmorphie) (7) |
- Lidachsen nach außen schräg ansteigend - Epikanthus (Sichelförmige Hautfalte am inneren Randwinkel des Auges (auch als Mongolenfalte bekannt)) - Brushfield-Flecken - Große Zunge - Brachyzephalus - Hypoplastischer Nasenknochen, Breite Nasenwurzel - Anomalien der Ohrmuschel (klein, rund, adhärente Ohrläppchen) |
|
|
Trisomie 21 (Down-Syndrom) ・Merkmale der Extremitäten, des Weichteilgewebes und des Skeletts (3) |
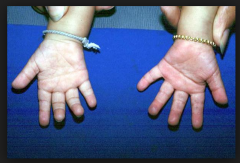
- Vierfingerfurche: Beugefalte in der Handinnenfläche, welche senkrecht zu den Fingern entlang der Fingergrundgelenke läuft - Sandalenfurche: Großer Zwischenraum zwischen 1. und 2. Zehe - Brachydaktylie (Kurzfingrigkeit) |
|
|
Trisomie 21 (Down-Syndrom) ・Organfehlbildungen (5) |
- Herzfehler in ca. 50% der Fälle (ASD, VSD, atrioventrikulärer Septumdefekt, Fallot-Tetralogie) - Duodenalstenose, Pancreas anulare, Analatresie - verminderte Fertilität bei Männern - Infektanfälligkeit - Leukämie-Risiko erhöht (ALL, AML) |
|
|
Trisomie 21 (Down-Syndrom) ・Körpergröße und Entwicklung (3) |
- Verzögerte motorische Entwicklung - Kleinwuchs (Körpergröße im Erwachsenenalter durchschnittlich 150cm) - Unterschiedliche Ausprägung der geistigen Retardierung (IQ im Mittel 50) |
|
|
Trisomie 21 (Down-Syndrom) ・Diagnostik (2) |
- Pränataldiagnostik zur Früherkennung - Postnatale Diagnostik |
|
|
Trisomie 21 (Down-Syndrom) ・Pränataldiagnostik zur Früherkennung (3) |
- Feinultraschall, Doppler-Sonographie - Invasive Untersuchungen zur Karyotypisierung - Tests aus mütterlichem Blut (va Triple-Test) |
|
|
Trisomie 21 (Down-Syndrom) ・Feinultraschall, Doppler-Sonographie (2) |
- Nackentransparenzmessung (Verbreiterung, breites dorsonuchales Ödem) - Nasenbeinmessung, Fetometrie |
|
|
Trisomie 21 (Down-Syndrom) ・Invasive Untersuchungen zur Karyotypisierung (3) |
- Chorionzottenbiopsie → Aussagekräftig und früh möglich (ab 10. SSW) - Amniozentese (ab 15. SSW) - CAVE: Abortrisiko 1-5% |
|
|
Trisomie 21 (Down-Syndrom) ・Triple-Test (3) |
- Durchführung während der Schwangerschaft (zwischen 15. und 18. SSW) möglich, aber keine Frühdiagnose! - Hinweise für Trisomie 21: Freies Östriol↓, AFP↓ und β-HCG↑ (um die 15. SSW) - Häufig falsch-positiv, daher umstritten |
|
|
Trisomie 21 (Down-Syndrom) ・Postnatale Diagnostik (2) |
- Chromosomen-Analyse! - Echokardiographie zum Nachweis von Herzfehlern (in 50%) |
|
|
Trisomie 21 (Down-Syndrom) ・Prognose (3) |
- Verminderte Lebenserwartung durch Organfehlbildungen, erhöhte Infektanfälligkeit und höheres Risiko für eine akute myeloische Leukämie - Nur etwa 45% der Patienten werden älter als 60 Jahre. - Die meisten Patienten mit Trisomie 21 entwickeln bis zum 40. Lebensjahr Zeichen der Alzheimer-Krankheit (Das Amyloid-Precursor-Protein, aus dem das β-Amyloid entsteht, liegt auf dem Chromosom 21) |
|
|
Gonosomale Chromosomenaberrationen ・Übersicht (2)
|
- Klinefelter-Syndrom → Ca. 1:800 (Korrelation mit dem Alter der Mutter) - Monosomie 45 (X0): Ullrich-Turner-Syndrom → Häufigkeit: 1:2500-3000 (Keine Korrelation mit dem Alter der Mutter) |
|
|
Klinefelter-Syndrom ・Karyotyp (2) |
- 47 XXY - selten 48XXXY und 48XXYY |
|
|
Klinefelter-Syndrom ・Klinik (6) (Bild) |
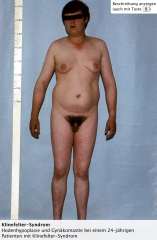
- Männlicher Phänotyp, wobei sich im Kindesalter kaum Aufälligkeiten zeigen. - Mit Eintritt in die Pubertät kommt es aufgrund des Testosteronmangels zu folgenden Symptomen → Eunuchoider Hochwuchs → Hodenhypoplasie und Hodendysgenesie bei normaler Penisgröße → Verminderte Fertilität, häufig Azoospermie → Gynäkomastie → Gehäufte Osteoporose im Erwachsenenalter durch Testosteronmangel |
|
|
Klinefelter-Syndrom ・Therapie (1) |
- Lebenslange Testosteronsubstitution (auch als Schutz gegen Osteoporose) |
|
|
Ullrich-Turner-Syndrom ・Karyotyp (1) |
- 45 XO |
|
|
Ullrich-Turner-Syndrom ・Klinik (8) (Bild) |

- Weiblicher Phänotyp - Lymphödeme an Hand- und Fußrücken bei Geburt - Geringes Längenwachstum - Ovariendysgenesie (Streak-Gonaden) → Östrogen- und Gestagenmangel (Primäre Amenorrhö, Infertilität) - Organfehlbildungen (Herzfehler , Fehlbildungen an Nieren und Harnleitern) - Pterygium colli (pathognomische Seitliche Halsfalten zwischen dem Processus mastoideus und dem Akromion) - Keine Intelligenzminderung - Normale Lebenserwartung |
|
|
Streak-Gonaden ・Definition (2) |
- Durch die fehlende Entwicklung der Ovarien aus den Keimzellen kommt es zu einer bindegewebigen Durchsetzung der Keimanlagen. - Streak-Gonaden können keine Hormone produzieren |
|
|
Ullrich-Turner-Syndrom ・Diagnostik (1) |
- Hypergonadotroper Hypogonadismus (Östrogene↓, FSH,LH↑) |
|
|
Ullrich-Turner-Syndrom ・Therapie (1) |
- Östrogen- und Gestagensubstitution |
|
|
- Weibliche Gonadendysgenesien ohne numerische Chromosomenaberrationen ・Übersicht (2)
|
- Swyer-Syndrom - Reine Gonadendysgenesie 46,XX |
|
|
Swyer-Syndrom ・Karyotyp (1) |
- 46 XY - Genetischer Defekt auf dem SRY-Gen → Kein Anti-Müller-Hormon → Keine Entwicklung der männlichen Geschlechtsorgane, aber Entwicklung von Uterus und Vagina (trotz XY-Chromosomensatz) |
|
|
Swyer-Syndrom ・Klinik (3) |
- Weiblicher Phänotyp - Bis zur Pubertät: Weibliche Entwicklung ohne Hinweise auf eine Chromosomenaberration - Ab der Pubertät: Östrogenmangel aufgrund der fehlenden Ovaranlage (Streak-Gonaden) → Primäre Amenorrhoe → Infertilität |
|
|
Swyer-Syndrom ・Therapie (1) |
- Östrogen- und Gestagensubstitution |
|
|
Reine Gonadendysgenesie ・Karyotyp (2) |
- 46 XX - Verschiedene Mutationen führen zu einer mangelhaften Entwicklung der Ovarien oder einem frühzeitigen Versiegen der Ovarialfunktion → Normale Entwicklung von Uterus und Vagina → Mangelhafte Östrogensekretion → Ausbleiben der Entwicklung sekundärer Geschlechtsmerkmale und Amenorrhö |
|
|
Reine Gonadendysgenesie ・Klinik (4) |
- Weiblicher Phänotyp - Bis zur Pubertät: Normale Entwicklung des weiblichen Genitale - Ab der Pubertät: Östrogenmangel aufgrund der mangelhaften Entwicklung der Ovarien oder des frühzeitigen Versiegens der Ovarialfunktion - Mangelnde Ausreifung der sekundären Geschlechtsmerkmale → Primäre (seltener: sekundäre) Amenorrhö → Infertilität |
|
|
Reine Gonadendysgenesie ・Therapie (1) |
- Östrogen- und Gestagensubstitution |