![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
91 Cards in this Set
- Front
- Back
|
Kollagenosen ・Definition (2) |
- Gruppe chronisch-entzündlicher Systemerkrankungen, die sich vorwiegend am Bindegewebe, an der quergestreiften Muskulatur und den Gefäßen manifestieren. - Große Bedeutung haben Kollagenosen wegen ihrer teilweise schweren Ausprägung an den inneren Organen. |
|
|
Kollagenosen |
- unbekannt - genetische Disposition (Assoziation mit bestimmten HLA-Antigenen) |
|
|
Kollagenosen |
- polyklonale B-Zell-Aktivierung → Durchbrechung der immunologischen Toleranz mit Bildung verschiedener Autoantikörper |
|
|
Kollagenosen |
- ANA (Antikörper gegen Zellkernantigene) - ENA (Antikörper gegen extrahierbare nukleäre Antigene) - SS-A (Antikörper gegen zytoplasmatische Glykoproteine) - SS-B (Antikörper gegen zytoplasmatische RNA-Protein) - Rheumafaktor (Antikörper gegen Immunglobuline) |
|
|
Spezifische Autoantikörper bei verschiedenen Kollagenosen (Tabelle) |

|
|
|
Kollagenosen |
- Systemischer Lupus erythematodes (SLE) - Progressive systemische Sklerose (PSS) - Polymyositis (PM) und Dermatomyositis (DM) - Sjögren-Syndrom - Mischkollagenose (Sharp-Syndrom) |
|
|
Systemischer Lupus erythematodes (SLE) ・Definition (1) |
- generalisierte, entzündliche Systemerkrankung, die durch das Auftreten charakteristischer Autoantikörper und Bildung von Immunkomplexen gekennzeichnet ist. |
|
|
Systemischer Lupus erythematodes (SLE) |
- genetische Disposition (HLA-Antigen DR2 und DR3) |
|
|
Systemischer Lupus erythematodes (SLE) |
- Verschiedene exogene und endogene Auslöser → polyklonale B-Zell-Aktivierung → gestörten Immuntoleranz mit Bildung von Autoantikörpern → über 2 verschiedene Mechanismen zu einer Schädigung der Zielstrukturen → 1. direkte antikörpervermittelte Zelllyse ↳ Bindung der Autoantikörper an die Zielstruktur (Opsonierung) ↳ direkte Zerstörung der Zellmembran mit anschließender Phagozytose (Typ-II-Reaktion nach Coombs und Gell) → 2. indirekte Gewebeschädigung durch Immunkomplexbildung ↳ subendotheliale Ablagerung der Immunkomplexe ↳ Komplementaktivierung ↳ Einstrom von Entzündungszellen (Typ-III-Reaktion nach Coombs und Gell) |
|
|
Systemischer Lupus erythematodes (SLE) |
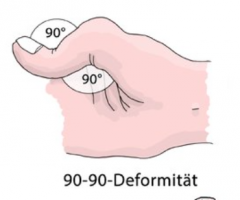
- Allgemeinsymptomen (Abgeschlagenheit, Fieber, Müdigkeit und Gewichtsabnahme) - Arthralgien - Myositis
- nicht destruierende Oligo- bzw. Polyarthritis (Z-Daumen (90°-90°-Deformität)) - Hautbefall v.a. an lichtexponierten Stellen (Schmetterlingserythem) - (diskoidale) papulöse Effloreszenzen, Schuppenbildung (Streckseiten der Finger, nicht ablösbar!) - periungualen Teleangiektasien - diffusen Alopezie (reversibel)
|
|
|
Systemischer Lupus erythematodes (SLE) |
- Perikarditis/Pleuritis (insbes. abakterielle Endokarditis Libman-Sacks) - Glomerulonephritis vom Immunkomplextyp (nephrotisches Syndrom) - 50 % Intimaproliferationen und Mikrothrombosierungen der kleinen Hirngefäße - Panzytopenie - sekundäres Antiphospholipidsyndrom ( Autoantikörpern gegen Phospholipide → rezidivierende Aborte, rezidivierende venöse/arterielle Thrombosen und Thrombozytopenie.) |
|
|
Systemischer Lupus erythematodes (SLE) |
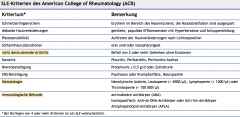
|
|
|
Systemischer Lupus erythematodes (SLE) |
- Labor - immunologische Diagnostik - Radiologie - Biopsie |
|
|
Systemischer Lupus erythematodes (SLE) |
- ↑Entzündungsparameter - polyklonalen Hypergammaglobulinämie (↑α2-Globuline und γ-Globuline) - ↓Komplementkonzentration (Abnahme der Komplementfaktoren C3, C4 und der gesamthämolytischen Aktivität CH50 - Leukozytopenie (zusätzlich evtl. Anämie und/oder Thrombozytopenie)
|
|
|
Systemischer Lupus erythematodes (SLE) |
- Nachweis von Autoantikörpern gegen Doppelstrang-DNA (ds-DNA-Autoantikörper, pathognomonisch) |
|
|
Systemischer Lupus erythematodes (SLE) |
- Röntgenaufnahme - Diagnose von Organkomplikationen (Arthrosonografie, CT, Angio, etc) |
|
|
Systemischer Lupus erythematodes (SLE) |
- Typisch für die nicht destruierende Arthritis sind (Sub-)Luxationen, die v.a. die kleinen Gelenke betreffen (sog. Jacoud-Arthritis) |
|
|
Systemischer Lupus erythematodes (SLE) |
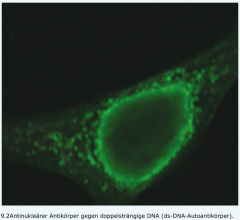
- bioptische Sicherung durch die feingewebliche Untersuchung betroffener Organe (v.a. Niere und Haut, Knochenmark) - fluoreszenzmikroskopisch granuläre Ablagerungen von Immunglobulinen und Komplementfaktoren an der Basalmembran im Bereich der dermoepidermalen Grenze bzw. Glomerula („Lupusband“)
- Nachweis auch in makroskopisch unbefallener, nicht lichtexponierter Haut (DD chronisch-diskoidale LE und kutan-disseminierte LE) |
|
|
Systemischer Lupus erythematodes (SLE) |
- „Lupus-like-Syndrom“ (eindeutige klinische Symptomatik, aber fehlende Immunphänomene) - medikamenteninduzierter Lupus erythematodes (Anti-Histon-Antikörper vorhanden, pathognomonische Anti-dsDNA-Antikörper fehlen.) |
|
|
DD Systemischer Lupus erythematodes (SLE) vs. kutaner Lupus erythematodes (chronisch-diskoider LE, CDLE) vs. subakut-kutaner Lupus erythematodes (SCLE) (Tabelle) |
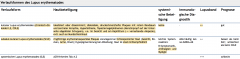
|
|
|
Systemischer Lupus erythematodes (SLE) |
- Pharmakotherapie |
|
|
Systemischer Lupus erythematodes (SLE) |
- Akuter Schub → hoch dosiert Steroide
- Milder Verlauf → Dauertherapie mit (Hydroxy-)Chloroquin (regelmäßige ophthalmologische Kontrollen) → bedarfsorientiert NSAR
- progressiver Verlauf → steroideinsparende Therapie mit Immunsuppressiva (Azathioprin, Methotrexat (va Gelenksbefall), Belimumab)
- schwere Verlaufsformen → Cyclophosphamid-Pulstherapie → Mycophenolatmofetil (MMF) (va Lupusnephritis)
- schwerste Verläufe → hoch dosierte Immunglobuline und Plasmapherese
- Antiphospholipidsyndrom → lebenslange Antikoagulation |
|
|
Exkurs: SLE und Schwangerschaft (3)
|
- hormonellen Umstellungen → deutliche Verschlechterung der Symptomatik (insb. Lupusnephritis) - ↑Abortrisiko - Möglichkeit des neonatalen Lupussyndrom (insb. bei SS-A-Autoantikörper) → Neugeborene mit Hauteffloreszenzen und Kardiomyopathie (Herzrhythmusstörungen) |
|
|
Progressive systemische Sklerose (PSS) ・Synonyme (1) |
- Sklerodermie |
|
|
Progressive systemische Sklerose (PSS) |
- generalisierte, entzündliche Systemerkrankung, die durch eine überschießende Kollagenbildung charakterisiert ist und zur Fibrosierung und Sklerosierung der Haut sowie der inneren Organe führt. - An den Gefäßen kommt es zu einer obliterierenden Angiopathie mit der Gefahr ischämischer Organinfarkte. |
|
|
Progressive systemische Sklerose (PSS) |
- ♂1:10♀ |
|
|
Progressive systemische Sklerose (PSS) |
- genetische Disposition HLA-DR1, -4, -5 und -8 |
|
|
Progressive systemische Sklerose (PSS) |
- gestörte Aktivierung des Immunsystems → zytokininduzierte Fibroblastenproliferation und gesteigerte Kollagensynthese → Fibrosierung und Sklerosierung des Bindegewebes → Gewebeinduration und Entzündungsreaktion mit Intimaproliferation. |
|
|
Progressive systemische Sklerose (PSS) |
- sekundäres Raynaud-Syndrom (häufigstes Frühsymptom, oft Jahre voraus) - Haut- und Gelenkbefall - Beteiligung innerer Organe - 2 Verlaufsformen → limitierte kutane Sklerodermie → diffuse Sklerodermie |
|
|
Progressive systemische Sklerose (PSS) |
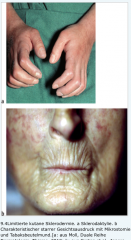
- Verläuft klassischerweise in 3 Stadien → 1. schmerzloses Ödem an Akren und distalen Extremitäten → 2. Sklerodaktylie, Madonnenfinger, Gesichtsstarre und Tabaksbeutelmund → 3. Atrophie der Haut, Pigmentverschiebungen, Teleangiektasien, digitalen Ulzerationen („Rattenbiss“ an Fingerkuppen) und nicht destruierende Arthritis |
|
|
Progressive systemische Sklerose (PSS)
|
- Schluckstörungen und Refluxösophagitis (Wandstarre des Ösophagus) - Malabsorption, Diarrhöe, paralytischer Ileus - pulmonaler Hochdruck und Cor pulmonale - progrediente Niereninsuffizienz mit einem renalen Hypertonus (eher selten) |
|
|
Limitierte kutane Sklerodermie ・Klinik (4) |
- Hautveränderungen an den distalen Extremitäten (unterhalb des Ellenbogens) und Akren (Sklerodaktylie); evtl. Gesichtsbefall (Mikrostomie) - Ösophagusstarre - Lungenfibrose - pulmonal-arterielle Hypertonie |
|
|
Diffuse Sklerodermie ・Klinik (2) |
- rascher und progressiver Befall des gesamten Integuments mit Ödem, Induration und Atrophie - regelmäßige Beteiligung innerer Organe (am häufigsten: Ösophagus, Lunge, Niere) |
|
|
Progressive systemische Sklerose (PSS) |
- Nagelfalzmikroskopie - Labor - Immunologische Diagnostik - Radiologie |
|
|
Progressive systemische Sklerose (PSS) |
- typische Gefäßkaliberschwankungen mit Stenosen und Aussackungen (sog. Megakapillaren) und eine Zunahme avaskulärer Felder (morphologisches Korrelat der funktionellen Gefäßspasmen) → > 90 % bereits in Frühphase |
|
|
Progressive systemische Sklerose (PSS) |
- ↑Entzündungsparameter - ↑ γ-Globuline |
|
|
Progressive systemische Sklerose (PSS) |
- Spezifisch bei diffuser Sklerodermie → Anti-Topoisomerase-Antikörper (Anti-SCL70) → Assoziation mit HLA-DR5
- Spezifisch bei limitierter kutaner PSS → Anti-Zentromer-Antikörper → Assoziation mit HLA-DR1, -4, -8 |
|
|
Progressive systemische Sklerose (PSS) |
- Röntgen - Sono - Biopsie - HR-CT, Ösophagusbreischluck, Echokardiographie (zur Diagnose der Organbeteiligung) |
|
|
Progressive systemische Sklerose (PSS) |
- Veränderungen an den Fingern → Akroosteolysen → Kalkablagerungen (Calcinosis cutis) → Spindelform der Endphalangen (wie „abgelutscht“) - Röntgen-Thorax-Aufnahme: Nachweis einer Lungenfibrose |
|
|
Progressive systemische Sklerose (PSS) |
- Nachweis von Kalkablagerungen |
|
|
Progressive systemische Sklerose (PSS) |
- obliterierende Angiopathie v.a. kleiner Blutgefäße, die hyalinisieren und veröden („Zwiebelschalenangiopathie“). |
|
|
Progressive systemische Sklerose (PSS) |
- Lunge: Gerüstfibrose - häufig auch Milchglasinfiltrate als Hinweis auf eine Alveolitis. |
|
|
Progressive systemische Sklerose (PSS) |
- primäres Raynaud-Syndrom - eosinophile Fasziitis (Morbus Shulman) - okalisierte (zirkumskripte) Sklerodermieformen - nephrogene fibrosierende Dermatopathie (bei dialysepflichtigen Patienten vermutlich durch das Kontrastmittel Gadolinium ausgelöst) |
|
|
eosinophile Fasziitis (Morbus Shulman) ・Definition (2) |
- Schwellungen und Induration der proximalen Extremitäten durch Fasziitis - DD Labor |
|
|
okalisierte (zirkumskripte) Sklerodermieformen ・Definition (4) |
- Betrifft ausschließlich Haut - lilafarbenen Ringen - Hände sind im Vergleich zur limitierten kutanen Sklerodermie nicht betroffen - Sonderform: Sklerodermie en Coup de Sabre (Sklerodermieherd aus, der typischerweise „säbelhiebartig“ vom Kapillitium zur Stirn zieht) |
|
|
Progressive systemische Sklerose (PSS) |
- Kälteschutz bei Raynaud-Syndrom - physiotherapeutische Bewegungsübungen - Pharmakotherapie |
|
|
Progressive systemische Sklerose (PSS) |
- Akuter Schub → Glukokortikoide
- Beteiligung innerer Organe → Dauertherapie mit Methotrexat oder Azathioprin
- fibrosierende Alveolitis → Cyclophosphamid-Pulstherapie |
|
|
Progressive systemische Sklerose (PSS) |
- chronisch progredient - imitierte kutane Sklerodermie hat eine deutlich bessere Prognose. |
|
|
Polymyositis (PM) |
- chronisch-entzündliche Systemerkrankung, die mit einer lymphozytären Infiltration der quergestreiften Muskulatur einhergeht |
|
|
Dermatomyositis (DM) ・Definition (2) |
- Polymyositis mit zusätzlich entzündlichen Veränderungen der Haut - 1/3 der PM-Patienten |
|
|
Polymyositis (PM) und Dermatomyositis (DM) ・Epidemiologie (4) |
- PM > DM - ♂1:2♀ - bevorzugt im 5.–6. Lebensjahrzehnt - < 20 % Beginn im Kindersalter (juvenile Dermatomyositis) |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- autoimmunologische Formen - paraneoplastische Form |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- idiopathische PM/DM |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- in 3-6 % PM/DM als paraneoplastisches Syndrom - Häifige Primärtumoren → Malignome des Intestinaltraktes → Malignome des Respirationstraktes → MammaCA → OvarienCA |
|
|
Polymyositis (PM) |
- schleichende, symmetrische Muskelschwäche v.a. proximaler Extremitäten im Bereich des Schulter- und Beckengürtels (Treppensteigen, Tragen von Gegenständen, Aufstehen aus sitzender Haltung, Muskelkater) - Typische Beteiligung der Nacken- und Schlundmuskulatur (Schlucken, Sprechen und Atmen) - Arthralgien - Raynaud-Phänomen - Beteiligung der inneren Organe (va Ösophagus und Myokard) |
|
|
Dermatomyositis (DM) ・Zusätzliche Klinik (4) (Bild) |
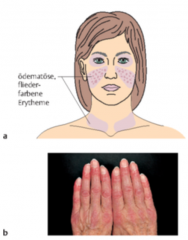
- symmetrisches lilafarbenes Erythem an Stirn, Wangen und Augenlidern (bei starker Ausprägung auch Hals, Dekolleté, Schultern und oberer Rücken) - charakteristischer trauriger Gesichtsausdruck (Facies myopathica) - erythematöse, schuppende Plaques an der Dorsalseite der Fingergelenke („Gottron-Zeichen“) (seltener an den Ellbogen, Kniegelenken und Knöcheln) - Atrophien und Hämorrhagien am Nagelfalz. |
|
|
Jo-1-Antikörpersyndrom ・Klinik (6) |
- Myositis - fibrosierende Alveolitis - Fieber - Raynaud-Symptomatik - Polyarthritis - Rhagaden an den Händen. |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- Labor - Immunologische Diagnostik - Elektromyografie (EMG) - Radiologie - Biopsie - Ausschluss eines malignen Tumors (paraneoplastische PM/DM) |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- ↑Entzündungsparameter - ↑Myoglobin - ↑Muskelenzyme (CK, GOT, Aldolase und LDH) |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- 40% Anti-Jo-1-Antikörper - 20 % Anti-PM-Scl-Antikörper (v.a. bei Overlap-Syndrom) - 10 % Mi-2-Autoantikörper (deuten auf eine DM hin) - 5–10 % Anti-SRP-Antikörper (deuten auf akuten Krankheitsverlauf mit Herzbeteiligung) |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- Fibrillationen (Denervierungszeichen) - frühe Rekrutierung motorischer Einheiten mit vollem Interferenzmuster bei nur leichter Muskelanspannung. |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- MRI |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- ödematösen Veränderungen im Bereich des betroffenen Muskels - hilft zur Lokalisation der geeigneten Biopsiestelle. |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- lymphozytäre Infiltration mit CD8-positiven T-Zellen - Faserdegeneration im Bereich der betroffenen Muskelfasern - DM spezifisch → Muskelfasern unregelmäßigen Kalibers → vermehrte atrophe Muskelfasern an den Faszikelrändern (perifaszikuläre Atrophie) → gruppenförmige Muskelfaserregenerate → Muskelfasernekrosen in Resorption → perivasale und interstitielle lymphohistiozytäre Infiltrate |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- juvenilen Dermatomyositis (subkutane Kalzinose tritt häufiger und relativ früh auf) |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- Im akuten Schub → hoch dosierte Steroidgabe
- Langfristig → steroideinsparende immunsuppressive Therapie mit MTX oder Azathioprin
- Lungenbeteiligung (z.B. im Rahmen des Jo-1-Antikörpersyndroms) → Cyclophosphamid
- therapierefraktärer Verlauf → hoch dosierte Immunglobulinbehandlung und Plasmapherese |
|
|
Polymyositis (PM) und Dermatomyositis (DM) |
- chronisch-progredient |
|
|
Sjögren-Syndrom ・Definition (2) |
- Chronisch-entzündliche Erkrankung, die sich an den exokrinen Drüsen manifestiert. - Am häufigsten sind die Tränen- und Speicheldrüsen betroffen (Sicca-Syndrom). |
|
|
Sjögren-Syndrom |
- zweithäufigste Autoimmunerkrankung - ♂1:9♀ |
|
|
Sjögren-Syndrom |
- primäres Sjögren-Syndrom → Ursache unbekannt → Assoziation mit den HLA-Merkmalen DR2 und DR3
- sekundäres Sjögren-Syndrom → Tritt in Begleitung anderer Autoimmunkrankheiten auf („Overlap-Syndrom“) → weitere Assoziationen mit rheumtatoiden Arthritis, Vaskulitiden, primär biliären Zirrhose, Hepatitis C, Hashimoto-Thyreoiditis etc. |
|
|
Sjögren-Syndrom |
- Sicca-Syndrom → Xerostomie (Mundtrockenheit, Zungenbrennen, Schluckbeschwerden, Durstgefühl, Kariesanfälligkeit) → Xerophthalmie (Keratoconjunctivitis sicca) - evtl. Heiserkeit, chronischem Hustenreiz und Störungen der Sexualfunktion - Arthralgien - sekundäre Vaskulitis (häufig beobachteten) - 6% Entwicklung eines malignen Lymphoms |
|
|
Sjögren-Syndrom |
- ophthalmologische Untersuchungsmethoden - Labor - immunologische Diagnostik - Radiologie - Biopsie |
|
|
Sjögren-Syndrom |
- Schirmer-Test weist eine verminderte Tränensekretion nach - Spaltlampenuntersuchung: Nachweis einer Keratoconjunctivitis sicca |
|
|
Schirmer-Test ・Ablauf (6) |
- Schirmer I → Bestimmung der Reiz-Tränensekretion
- Schirmer II → Bestimmung der Basis-Tränensekretion |
|
|
Sjögren-Syndrom |
- polyklonale Hypergammaglobulinämie (→ Bei monoklonalen Hypergammaglobulinämie an Lymphome denken) - ↑BSG - Anämie, Leukopenie und Thrombozytopenie |
|
|
Sjögren-Syndrom |
- Autoantikörper gegen zytoplasmatische Proteine (SS-A und SS-B) - Autoantikörper gegen das Epithel der Speicheldrüsenausführungsgänge - 50 % Rheumafaktoren |
|
|
Sjögren-Syndrom |
- Sono - szintigrafische Untersuchung |
|
|
Sjögren-Syndrom |
- Glandula parotis zeigt wabig aufgelockertes Drüsenparenchym mit echoreichen Septen |
|
|
Sjögren-Syndrom |
- Nachweis einer verminderten Speichelsekretion |
|
|
Sjögren-Syndrom |
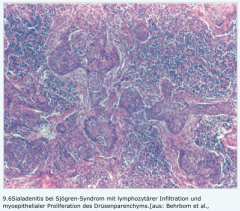
- Geeignete Entnahmeorte: Speicheldrüsen oder Lippeninnenseiten - Nachweis von infiltrativen Drüsenveränderungen (fokale Sialadenitis) mit Trias → lymphozytäre Infiltration → Parenchymatrophie → myoepitheliale Proliferation |
|
|
Sjögren-Syndrom
|
- primäres Sjögren-Syndrom → Keratoconjunctivitis sicca (Schirmer-Test und Spaltlampenuntersuchung) → positiver Biopsiebefund (fokale Sialadenitis mit Lymphozyteninfiltration)
- sekundäres Sjögren-Syndrom → Keratoconjunctivitis sicca & positiver Biopsiebefund → zusätzlich muss eine auslösende Grunderkrankung nachweisbar sein. |
|
|
Sjögren-Syndrom |
- Pharmakotherapie |
|
|
Sjögren-Syndrom |
- Cholinergika (zb Bromhexin, Pilocarpin) (symptomatisch zur Steigerung der Tränen- bzw. Speichelsekretion) - NSAR, ggf. in Kombination mit Chloroquin (bei Arthritiden) - Azathioprin oder MTX (nur bei Auftreten einer sekundären Vaskulitis und/oder viszeraler Organbeteiligung) |
|
|
Sjögren-Syndrom |
- i.d.R. gut - verschlechterung bei Beteiligung innerer Organe, sekundärer Vaskulitis oder Lymphomentwicklung - sekundären Sjögren-Syndrom abhängig von begleitender Grunderkrankung |
|
|
Mischkollagenose (Sharp-Syndrom) ・Synonyme (3) |
- Überlappungssyndrom „overlap-syndrome“ - gemischte Kollagenkrankheit - MCTD/UCTD = „mixed/undetermined connective tissue disease“ |
|
|
Mischkollagenose (Sharp-Syndrom) |
- Systemerkrankungen, die sich am Bindegewebe und Gefäßsystem abspielen und klinische Symptome verschiedener Kollagenosen zeigen. - Die häufigste Mischkollagenose ist das Sharp-Syndrom |
|
|
Sharp-Syndrom ・Definition (3) |
- klinischen Symptomatik von → SLE → Dermato-/Polymyositis → progressiven Systemsklerose |
|
|
Mischkollagenose (Sharp-Syndrom) |
- ♂<♀ - Manifestationsgipfel im 5. Lebensjahrzehnt |
|
|
Mischkollagenose (Sharp-Syndrom) |
- beginnt i.d.R. schleichend (Monate bis Jahre) - Beginn meist mit Raynaud-Syndrom - chronischen Polyarthritis (SLE) - progressiven systemischen Sklerose - Polymyositis - Beteiligung von Nieren, ZNS und Herz ist vergleichsweise selten. |
|
|
Mischkollagenose (Sharp-Syndrom) |
- Autoantikörpern gegen Ribonukleoprotein (Anti-RNP-Antikörper) (charakteristisch für das Sharp-Syndrom) |
|
|
Mischkollagenose (Sharp-Syndrom)
|
- Therapie entspricht den übrigen Kollagenosen - Prognose ist gut, - Übergang in klassische Kollagenose (insb. SLE) ist möglich. |