Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
12 Cards in this Set
- Front
- Back
|
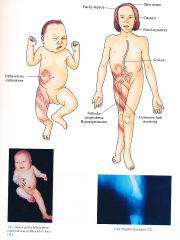
Clinical Conradi-Hunermann Syndrome/ X0linked dominant chondrodysplasia punctata; Conradi-Hunermann-Happle syndrome
|
|
|
|
Synonym
|
X linked dominant chondrodysplasia punctata; Conradi Hunermann Happle syndrome
|
|
|
Inheritance
|
X linked dominant; Emopamil binging protein (EBP) gene on Xp11
|
|
|
Prenatal
|
Ultrasound evaluation of long bones
|
|
|
Incidence
|
Rare; usually lethal in males; reports of surviving males both with/without 47, XXY
|
|
|
Age at Presentation
|
Birth
|
|
|
Pathogenesis
|
Mutation in the EBP gene or 3P hydroxysteroid A I_A7_ isomerase leads to a defect in cholesterol biosynthesis and can explain skeletal phenotype
|
|
|
Clinical
|
Skin
lchthyosiform erythroderma in Blaschko's lines in infancy; resolves with follicular atrophoderma and/or hyperpigmentation Hair Coarse, patchy alopecia Eyes Asymmetric focal cataracts Musculoskeletal Stippled epiphyses (punctate calcifications), asymmetric limb shortening, short stature, scoliosis Cranofacial Frontal bossing, macrocephaly, flat nasal root, asymmetric Central Nervous System Mental retardation (rare) |
|
|
D/Dx
|
Autosomal recessive rhizomelic chondrodysplasia punctata
X linked recessive chondrodysplasia punctata with steroid sulfatase deficiency CHILD syndrome Incontinentia pigmenti |
|
|
Lab
|
Bone films
Neonatal skin biopsy may reveal calcium in the epidermis with von Kossa's stain Peroxisomal function in cultured fibroblasts |
|
|
Management
|
Referral to orthopedist, dermatologist, ophthalmologist
Examine first degree relatives |
|
|
Prognosis
|
Ichthyosis and stippled epiphyses resolve after infancy; orthopedic complications
predominate with normal life span |