Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
137 Cards in this Set
- Front
- Back
|
What differs anemia and anemic syndrome?
|
Anaemia= less than the normal quantity of hemoglobin in the blood
Anaemic syndrome =clinical syndrome caused by tissue hypoxia |
|
|
Normal value of HB and HTK?
|
HB 136-176 men (120-170 women)
HTK 0,38-0,48 |
|
|
What do they stand for?
RBC MCV MCH MCHC |
RBC= no. of ery
MCV= Volume of ery MCH= Mass of HB in ery MCHC= HB cooncentration in ery |
|
|
What are the symptoms of anic syndrome?
Primary, compensation ans secondary? |
Primary: Tissue hypoxia: pallor, fatigue, weakness, dyspnea
Compensation and adaptation: Hypercinetic circulation, palpitations, tinnitus Secondary : Cardiovascular symptoms – decompensation of ischemic heart disease, AP, IM, claudications |
|
|
Progress and severity of AS depends on:
|
1. Absolute value of Hb
Hgb 70-80 g/l = most of patients suffer from symptoms 2. Speed of onset 3. Age and overall performance of the patient |
|
|
ANAEMIA - CLASSIFICATION
acc. to MCV |
Morfologic criteria:
According to MCV: (80 – 95 fl) microcytic, normocytic, macrocytic |
|
|
ANAEMIA - CLASSIFICATION
acc. to MCH |
According to MCH: (27-32 pg)
normochrome, hypochrome |
|
|
MICROCYTIC ANAEMIA
4 examples |
MICROCYTIC ANAEMIA
Iron deficiency anaemia Chronic disease anaemia Thalasemia, sideroblastic anaemia |
|
|
MACROCYTIC ANAEMIA
2 types + reason |
MACROCYTIC ANAEMIA
Megaloblaste anaemia(lack of B12, folic acid, MDS) Macrocytic non-megaloblaste anaemia (usually secondary |
|
|
NORMOCYTIC ANAEMIA
Primary imparement |
Primar impairment of blood marrow: aplastic anaemia, MDS, PNH,
myelofibrosis. |
|
|
NORMOCYTIC ANAEMIA
Secondary impairmen |
Secondary impairment of blood marrow :(infiltration, infection,
endocrinological and systemic diseases, ACD) |
|
|
. ANAEMIA WITH RETICULOSIS
|
Haemolytic aneamia corpuscular
Haemolytic anaemia extracorpuscular – imunne and non-immune based |
|
|
Pathofysiological classification of anemia
|
Proliferation and differentiation disorder
Increased destruction of RBC Blood loss Combined etiology |
|
|
Where does iron defic. and ACD affect hb synth?
|
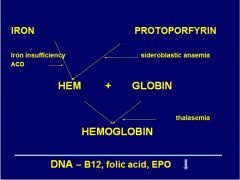
|
|
|
Where does sideroblastic anemia affect Hb synth?
|
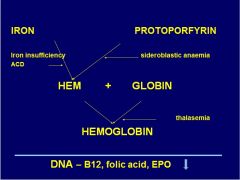
|
|
|
Where does thalasemia affect hb synth?
|
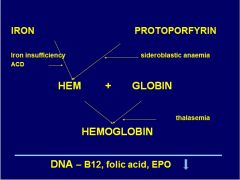
|
|
|
Where is B12, folic acid and epoxide an importnant factor in hb synth?
|
DNA synth
|
|
|
Which anemia is the most common? (80%)
|
Iron deficiency
|
|
|
Sideropenia=?
Happens in how many fertile women? |
Iron deficiency
35-58% of fertile women |
|
|
What does iron defic. affect except for blood count? (6)
|
DNA synthesis impairement
Tissue fosforylation impairement Purine metabolism impairement Colagen synthesis impairement Granulocyte function impairement Neurotransmiter function impairement |
|
|
How is iron stored in the body? How many %, where?
|
Hb: 70%
Ferritin: 25% Myoglobin:4% Enzymes: 1% |
|
|
What is the interesting thing with iron excretion?
|
There is no physiological excretion mechanism!
|
|
|
How much iron do we absorb every day? (and excrete)
And how much does the diet contain? |
1-2 mg is absorbed even if the diet contains 10-20 mg.
|
|
|
Where does most of the iron absorbtion occur?
|
Duodenum and upper part of jejunum
|
|
|
How much iron supply do we have in the organism?
|
3000-5000 mg
|
|
|
What is hepicidin?
Come from, stimulatet by? What does it do? |
Hepcidine
Acute phase reactant Source: hepatic cells, heart, Iron stimulates Hepcidine Hepcidine inhibits iron absorption in the intestine, iron release from macrophages and iron transport via placenta ACD, hereditary hemochromatosis |
|
|
How much iron is absorbed if you compare meat and vegetables?
|
Meat: 25-30% of iron is absorbed
- Vegetables: 5% of iron is absorbed So vegetariaans are in risk for iron defic. |
|
|
What happens with 65% of pat with stomach resection?
|
They get aabsorbtion disorders. Can lead to iron defic.
|
|
|
How much iron do you lose during menses?
|
ca 3mg/day
|
|
|
When is there an increased need for iron?
|
Gravidity
Brest-feeding Growth |
|
|
Symptoms of iron defic.
What do they mean? |
Anemic syndrome
Cefalea, paresthesia, fatigue Tongue burning, angulitis Odyno-(painful), dysfagia Sy Kelly-Patterson (hair, nails) Brittle hair, nails (Pica, pagofagia) |
|
|
Physical examination findings when iron defic.
|
Pallor – skin, mucous membrane
Blue sclerae Ulcers/ angulitis Smooth tongue Straight/(spoon-shaped) nails Achlorhydria (low production of hcl), atrophic gastritis |
|
|
Laboratory findings in iron def. 1
|
RDW (rbc distribution): high
Trombocytosis (over 50% of patients) BM –staining for iron - lack of Fe in siderophages - sideroblasts lower then 10% |
|
|
Laboratory findings in iron def. 2
MCV MCH MCHC TRanferrin S-transferrin transferrin satur. S-sTfr |
MCV under 80fl
MCH under 25ug MCHC – late symptom Transferrin -increased S-ferritin <20ug/l Transferrin satur. – under15 % (N: 20-40%) VKFe (TIBC): increased S-sTfR > 8g/l |
|
|
Diff. diagnose microcytic anemia:
IRON INSUFF. Fe TIBC satTRF ferritin TRF receptor |
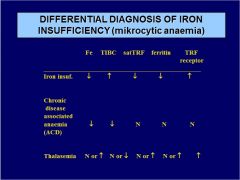
|
|
|
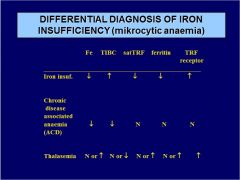
Diff. diagnose microcytic anemia:
ACD Fe TIBC satTRF ferritin TRF receptor |
|
|
|
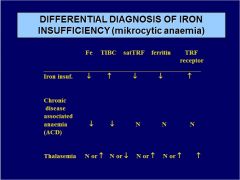
Diff. diagnose microcytic anemia:
THALASEMIA. Fe TIBC satTRF ferritin TRF receptor |
|
|
|
What is ferritin?
|
Acute phase reactant
Nespecific tumorous marker Level increases with age (75ug/l in old people = ? = iron defficiency |
|
|
3 types of Iron defficiency:
|
Prelatent
Latent Manifest - SA |
|
|
Treatment of IDA
|
Treatment of the cause of iron loss
+ iron supply |
|
|
Ferrotherapy
|
150-200mg Fe / day
Until enough supply is formed (ferritin 50ug/l) Use on an empty stomach CAVE: polyphenols, milk, egg yolk Dyspepsia Parenteral forms (CAVE: anaphylaxis; x new forms are safer - karboxymaltose) |
|
|
If P.o iron treatment is insuff agains iron defic...?
|
Ineffective treatment:
1. Diagnosis checking :BM examination, GIT examination aso…. Cave self-harming 2Switch to i.v. therapy |
|
|
What is Thalassemia and which types are there? Which one is worse?
|
Inherited disorder of Hb structure:
α –thalasemia = α disorder β – thalasemia = β disorder β – thalasemia is worse Can also be devided into minor (usually no symptoms), intermedia (only in Beta) and major (from both parents) depending on ammount of inheritence. MICROCYTIC ANEMIA |
|
|
What is a typical skull manifistation in a anemic disease?
|
Diploetic fibers from the skull in thalassemia.
|
|
|
Lack of B12of folic acid, 2 reasons:
|
Lack of B12of folic acid:
1. Pernicious anaemia - B12 absorption in the distal intestine disorder due to lack of intrinsic factor (produced by parietal cells of gastric mucosa) 2. Dihydrofolat reductase inhibitors (MTX, ARA-C) |
|
|
MEGALOBLASTIC ANAEMIA -CAUSES
|
Insufficient intake of B12 of folic
Absorption impairement: Increased demand Increased loss (hepatic laesions, bleeding) dihydrofolat reductase inhibitors (MTX,pyrimethamin) Transport disorders because of lack of transkobalamin I. and II |
|
|
Megaloblastic anemia
Blood count: |
macrocytes (↑MCV, ↑MCH, normal MCHC),
↓RTC, megalocytes, megaloblasts, leukocytosis with left shift, thrombocytopenia |
|
|
Megaloblastic anemia
Bone marrow |
hyperplasia of erytropoiesis, megaloblasts
|
|
|
Megaloblastic anemia
Biochem |
↓ B12, ↓folic acid, ↑direct and indirect
bilirubin, Normal iron! |
|
|
Where is B12 absorbed?
|
B12 absorption in ileum, binding to specific receptor (cubilin)
|
|
|
TRANSCOBALAMIN I's role in B12 mtb
|
Binds B12 in plasma,binds to B12 in stomach before binding to intrinsic factor, produced by neutrofiles and cells with exocrine secretion, his lack leads to low serum B12 levels
|
|
|
TRANSCOBALAMIN II's role in b12 mtb
|
Enables B12 absorption by cells, receptor on all type of cells, produced by endotelial cells, fibroblasts, ileum cells.., his lack leads to severe B12 deficiency in cells
|
|
|
What 3 factors does b12 absorbtion need?
|
1. intrisic factor
2. Transcobalamin I 3. Transcobalamin II |
|
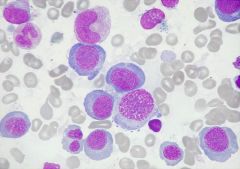
What is this?
|
Pernicious anaemia
|
|
|
Anaemia with increased no of reticulocytes + subtype
|
HEMOLYTIC ANAEMIA
- corpuscular - estracorspuscular bleeding - acute |
|
|
Corpuscular Haemolytic anaemias, pathogenesis
|
Lack of and defects in membrane proteins (ankyrin, spectrin, etc.)
leading to extravascular hemolysis (spleen) |
|
|
Herediatry spheracytisos is what?
Treatment? |
ongenital spherocytic anemia is a disorder of the surface layer (membrane) of red blood cells. It leads to red blood cells that are shaped like spheres, and premature breakdown of red blood cells
Treatment: Spleenectomy |
|
|
ERYTROCYTE ENZYMOPATHY
Diff. reasons? Type of anemia? |
CORPUSCULAR HEMOLYTIC ANEMIA
Defects in enzymes of anaerobe glykolysi (anemia with Heinz bodies) Defect in enzymes of pentos cycle |
|
|
GLUCOSE-6-PHOSPHATE DEHYDROGENASE defficiency
Results in what? How do we treat it? |
Results in: lack of NADPH …. Increased sensitivity to oxydating agents
Treatment: Prevention of exposure to oxydative agents (medication: antimalarics, sulfonamides,. Food: vicia fava etc.) , splenectomy, stem cell transplant |
|
|
sicle cell anemia – HbS, Thalassemia methHb and so on are example of?
|
Hemoglobinopathies
|
|
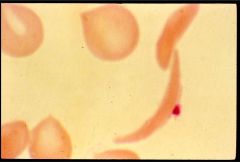
What is this?
|
Sickel cell anemia
|
|
|
Sicle cell anemia:
Pathogenesis |
Substitution glutamate valin on 6. position
chain: Hb polymerisation, deformation of erythrocyte, Shape=sickle cell. |
|
|
Sickle cell anemia:
Inheritance: |
Autosomal dominant type
homozygotic form – both B chains impaired heterozygotic form – one B chain impaired 25-50% HbS – sickle cell trait |
|
|
Sickel cell anemia:
What does it lead to? |
Hemolysis extravascular + intravascular (small vessel obstruction)
|
|
|
Sickle cell anemia:
Clinical picture treatment |
Hemolytic + aplastic crisis, splenomegaly
Treatment: Crisis prevention, transfusions, SCT |
|
|
Pathogenesis of AIHA:
|
Cooperation disorder among supresor T helper T lymphocytes and B lymphocytes responsible for immunity control
Dysregulation of this system leads to insufficient supression of antibody formation against own antigens |
|
|
HEAT ANTIBODIES
(AIHA) |
IgG character – optimal at 370C
Catch up of erythrocytes with binded antibody by spleen macrophages EXTRAVASCULAR HEMOLYSIS Activation of complement by high antibody titre INTRAVACULAR HEMOLYSIS |
|
|
COLD ANTIBODIES
(AIHA) |
IgM character – optimál at 40C
Bound to erytrocytes in colder acral parts, possibility of complement activation, ery aglutination INTRAVASCULAR HEMOLYSIS EXTRAVASCULAR HEMOLYSIS |
|
|
AIHA –laboratory parameters
Blood count Biochem Special test Proof of intravascular hemolysis? |
makrocytic anaemia with reticulocytosis
Biochemistry: direct and indirect bilirubin, urobilinogen in urine Special tests: Direct and indirect antiglobuline test (Coombs test) INTRAVASCULAR HEMOLYSIS PROOF: free Hb in plasma, levels of haptoglobin and hemopexin in serum, hemoglobinuria |
|
|
AIHA treatment
Light form ( Hb > 80 g/l ): SEVERE form ( Hb < 80 g/l ): |
Prednisone until Coombs test negative.
When therapy is ineffective: CYCLOPHOSPHAMIDE Severe: Both above combined + transfusion. |
|
|
What are the two main functions of the spleen?
Other functions? |
Filters blood to remove damaged/old RBC- red pulp
Serves as secondary lymphoid tissue by removing infectious agents and using them to activate lymphocytes- white pulp Other: A significant reservoir for T lymphocytes Plays an active role in the production of IgM antibodies and complement Has significant role in the functional maturation of antibodies |
|
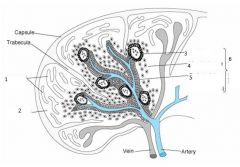
|
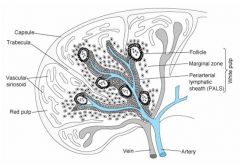
1. Vascular sinusoinds
2. Red pulp 3. follicle 4. Marginal zone 5. Periarterial lymphatic sheet (PALS) 6. White pulp |
|
|
Function of White pulp?
|
The white pulp is circular in
structure and is made up mainly of lymphocytes. It functions in a way similar to the nodules of the lymph node. |
|
|
Function of Red pulp?
|
The red pulp surrounds the white
pulp and contains mainly red blood cells and macrophages. The main function of the red pulp is to phagocytize old red blood cells |
|
|
When is it splenaomegaly (significant)?
|
When Palpable = significant, clear, usually clinically important. Usually after 1,5-2x increase in size.
|
|
|
Infections that cause splenomegaly?
|
Bacterial: Typhoid fever, endocarditis, septicemia, abscess
Viral:E-B virus, CMV, and others Protozoal: Malaria, toxoplasmosis |
|
|
Hematology benign disorders
that cause splenomegaly? |
Hemolytic anemia
Extramedullary hematopoiesis: thalassemia, osteopetrosis, (myelofibrosis |
|
|
Metabolic diseases that cause splenomegaly?
|
Lipidosis: Niemann-Pick, Gaucher disease
Mucopolysaccharidosis infiltration: Histiocytosis |
|
|
Other randoms that can cause splenomegaly?
|
Congestion (eg right heart failure, thrombosis of v. lienalis)
Cirrhosis Cysts + abscesses |
|
|
Causes of splenomegaly in malignant haematology (5)
|
Myeloprolipherative diseases (MPD)
2. leukemia 3. Lymphoma 4. Histocytic disease 5. Systemic mastocytosis |
|
|
Hypersplenism
|
Refers to a variety of ill effects resulting from increased splenic function that may be improved by splenectomy
Hypersplenism can be categorized as primary or secondary |
|
|
The criteria for diagnosis of hypersplenism included:
|
The criteria for diagnosis included:
Anemia, leukopenia, thrombocytopenia or a combination of the three Compensatory bone marrow hyperplasia Splenomegaly |
|
|
Felty’s Syndrome
|
Is a syndrome consisting of severe rheumatoid arthritis, granulocytopenia and splenomegaly
It usually occurs in patients with a long history of rheumatoid arthritis Severe, persistent and recurrent infections are characteristic Moderate splenomegaly is common Splenectomy is effective in most patients |
|
|
Infectious Mononucleosis in realation to splenomegaly
|
A disease characterized by fever, sore throat, lymphadenopathy and atypical lymphocytes
Most patients are young Clinical symptoms are similar to those of a severe upper respiratory tract infection The spleen is enlarged and palpable in over 50% of patients Splenic rupture may occur |
|
|
Diagnosis of splenomegaly
(Methods) |
Palpable vs non palpable
US and/or CT scan (PET) (to decide if homegenous or not) Crucial point: cause of splenomegaly |
|
|
Gaucher’s Disease
|
Is a disorder of lipid metabolism that may result in massive splenomegaly and hypersplenism
Commonly found in the Jewish population Diagnosis is made by finding the typical Gaucher’s cells in biopsy tissue Massive splenomegaly is usually the most common form of presentation The adult form is the most common form Splenectomy (subtotal) shows great benefits |
|
|
Blood Compositional Changes in the Asplenic or Hyposplenic Patient
|
The absence of functional splenic tissue results in characteristic changes in the circulating blood
Some of these are predictable and desirable results These changes are considered a measure of its success when splenectomy is performed for a hematologic disease Howell-Jolly bodies (nuclear remnants) and thrombocytosis (desired result) Other findings include: target cells, acanthocytes (spur cells), Heinz bodies (denatured hemoglobin) and stippled red cells |
|
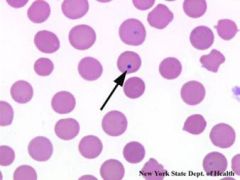
|
Howell Jolly bodies
Howell-Jolly bodies are round, purple staining nuclear fragments of DNA in the red blood cell |
|
|
Complications of splenectomy
|
Long life OPSI risk
Earleir onset of atherosclerosis |
|
|
Postsplenectomy Sepsis (OPSI)
|
Asplenic patients have an increased susceptibility to the development of overwhelming infection caused by encapsulated microorganisms.
The risk of sepsis is approximately 60 times greater than normal after splenectomy |
|
|
Postsplenectomy Sepsis
The most common bacteria: |
The most common bacteria: Streptococcus pneumoniae, Neisseria meningitidis or Haemophilus influenzae
|
|
|
What is a Differential count?
|
Seperation and counting the leukocytes
|
|
|
How many leu is there in peripheral blood/L?
|
4,0 – 10,0 x109/l
|
|
|
in which ''pools'' can we find the netruphils + their %?
|
Marrow pool: 90% neutrophils
Blood pool: 3% Tissue pool: 7% (eg mucosa) |
|
|
Where are Granulocytes & monocyte formed?
|
Granulocytes & monocytes are formed only in bone marrow
|
|
|
Lymphocytes are produced in?
|
Lymphocytes are produced in MB + in various lymphoid tissues (thymus, lymphnodes, splen, etc)
|
|
|
Granulocyte life span
|
Granulocytes:
after released from bone marrow, 4-8 hours circulate in blood & another 4-5 days in the tissues. Survive only for few hours in serious infection |
|
|
Monocytes lifespan:
|
10-20 hrs in blood.
Once in tissue they get much larger size to become tissue macrophage → life span month(s) |
|
|
Life span lymphocytes:
|
Life span for week or years depending on body’s need.
They continually circulate in blood & move from blood to tissues & from tissues to blood and again blood to tissues. |
|
|
How are the lobes in the neu connacted?
|
By chromatin
|
|
|
Arneth count
|
Arneth count: More the no. of lobes, the more mature is the neutrophil. More the no. of mature cells, Arneth count shifts to right (Vit. B12 or folate deficiency).
More the younger cells → shift to left (infection). |
|
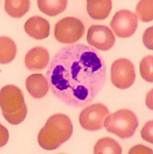
|
Neutrophil
|
|
|
Neutrophil's granules:
|
neutrally
stained granules 1. Primary/ lysosomal granules: less in no. containing acid hydrolases 2. Secondary granules: more numerous. Contain Lactoferin, |
|
|
Neutrophil function
|
Phagocytosis: 1st line of defence, ingest & destroy a bacteria.
Pyrogens & inflammatory cytokines: endogenous pyrogen which is an important mediator of febrile response to bacteria + other regulation and inflam. defense reaction |
|
|
Reactive neutrophilia example:
Physiological (6) Pathological (3) |
B. Pathological 1)Acute pyogenic (pus forming) infections, 2)Following tissue destruction
|
|
|
Neutropenia
Examples of reasons: (6) |
In children, 2) Typhoid, paratyphoid fever, 3) Viral infection, 4) Malaria, 5) Aplasia of bone marrow,
6) Bone marrow supprepression (failure). |
|
|
Leukemoid reaction
|
Non malignant excesive increase in leukocytes (in most cases neutrophils) in reaction to mostly inflammatory noxas.
WBC can increase up to 40-60 x 109/l , if neutrophilia significant left shift in differential is apparent. In most cases: sepsis (diffuse peritonitis, acute cholecystitis, acute pyelonephritis, etc.) |
|
|
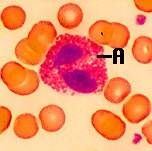
Eosinophil
Nucleus: Usually (85%) cells ‘bilobed Cytoplasm: Acidophilic, appears light pink in colour after staining 2. Granular |
|
|
Eosinopenia
Causes (3) |
Causes are:- 1) ACTH & steroid therapy, 2) Stressful conditions, & 3) Acute pyogenic infections
|
|
|
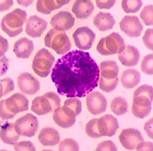
Basophil
Nucleus: irregular bilobed, often ‘S’ shaped & its boundary is not clear because of overcrowding with coarse granules. Cytoplasm: Is slightly basophilic & appear blue, it is full of granules. |
|
|
Mast cells
|
Large tissue cells resembling basophils. Present in bone marrow & immediately outside the capillaries in the skin.
These do not enter the blood circulation (normally). Functions: Mast cells play role in allergic reactions similar to the basophis. |
|
|
Monocyte granules:
|
Azur granules
|
|
|
CD56,CD57
|
NK cells
|
|
|
CD3
|
T cell rec
|
|
|
CD4
|
Thelper 0,1,2
|
|
|
CD8
|
Cytotoxic T cell
|
|
|
CD19,CD20
|
B lymphocyte
|
|
|
Pelger Huët´s
|
Pelger Huët´s abnormal leukocyte morphology:
autosomal dominant inher. disease: hyposegmentation of the nucleus |
|
|
May-Hegglin´s
|
May-Hegglin´s leukocyte abnormality: inherited together leukopenia + Döhle´s inclusions in neutrophils + thrombocytopenia with giant platelets.
|
|
|
Leukocyte adhesion defect (LAD):
|
autos. Recessive membrane defect due to lack of CD18 molecule. Defect in chemotaxis, aggregability and phagocytosis. Clinicaly: necrotic skin infections, pneumonits, otitis etc. Therapy: aloSCT.
|
|
|
autos. Recessive membrane defect due to lack of CD18 molecule. Defect in chemotaxis, aggregability and phagocytosis. Clinicaly: necrotic skin infections, pneumonits, otitis etc. Therapy: aloSCT.
|
autom. recessive disease with impaired migration and degranulaction of granulocytes. Comes with albinism, nystagmus, photophobia, mental retardation, frequent bacterial infections, peripheral neuropathy. Therapy: aloSCT
|
|
|
When do we see Döhle´s inclusions?
|
May-Hegglin's abnormality in neutrophils
|
|
|
How many healthy adults have palpable lymphnodes?
|
56%
|
|
|
In palpable LN, benign vs. malignant, percentage in individuals below 30 and above 50?
|
In individuals < 30 years: 80% benign etiology
In individuals > 50: 60% malignant etiology |
|
|
Assessing adenopathy: History
|
Age
Tumor or TBC in the history Allergy Drugs/medication Duration of adenopathy Associated complains (symptoms): fever, night swetting, malaise, weight loss, artralgy, sore throat, fatigue etc. Smoking Travels |
|
|
What to look for when doing phsyical examination when lymphadenopathy?
|
Location
Size Sensitivity consistency reaction of surrounding area |
|
|
Autoimmune disorders causing lymphadenopathy: (5)
|
RA, SLE, dermatomyositis, MCTD, Sjögren´s
|
|
|
Frequent causes of adenopathy according to the location
Inguinal (or axillar) adenopathy 1cm: |
Inguinal (or axillar) adenopathy 1cm: usually benign
|
|
|
Frequent causes of adenopathy according to the location
Cervical adenopathy: |
Cervical adenopathy: infections, carcinomas (consistency), lymphomas. Sialoadenitis (pseudolymphadenopathy)
|
|
|
Frequent causes of adenopathy according to the location:
Mediastinal adenopathy: |
Mediastinal adenopathy: lymphomas (mediastinum anterior), sarcoidosis, metastatic cancer
|
|
|
Frequent causes of adenopathy according to the location
Isolated axillar adenopathy: |
Isolated axillar adenopathy: infection, breast Ca, lymphoma
|
|
|
Frequent causes of adenopathy according to the location
Isolated inguinal adenopathy |
Isolated inguinal adenopathy (significant): infection (also veneral), lymphoma, metastatic Ca (consistency)
|
|
|
Frequent causes of adenopathy according to the location
Generalized adenopathy: |
Generalized adenopathy: infection (EBV, HIV, etc.), malignant lymphomas, CLL
|
|
|
Tender Vs. painless LN
|
lymph nodes that are tender are more likely to be due to an infectious process,
whereas painless adenopathy raises the concern of malignancy. |
|
|
Lymph node consistency
Lymphoma, ca, infectious? |
lymph nodes containing metastatic carcinoma are rock hard,
lymph nodes containing lymphoma are firm and rubbery, lymph nodes enlarged in response to an infectious process are soft. |
|
|
INdication for LN biopsy?
|
Constitutional symptoms (weight loss, fever, night sweats) otherwise unexplained
Persistent adenopathy > 4 - 6 weeks, otherwise unexplained Increasing size of the lymph node for several weeks Appearence of additional lymph nodes Abnormal blood test results (anemia, elevetad ESR, LDH, liver chemistries), otherwise unexplained Abnormal chest radiograph (e.g. mediastinal adenopathy) |
|
|
LN biopsy comments:
|
Excision of the whole lymh node strongly
preferred Reasonably experienced surgeon preferred Fine needle biopsy must be avoided |