Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
92 Cards in this Set
- Front
- Back
- 3rd side (hint)
|
Drug Action
|
Molecular action - invisible
|
|
|
|
Drug effect
|
pharmacologic effect- visible response
|
|
|
|
pharmacokinetics
|
time course of absorption, action, and elimination
|
|
|
|
pharmacodynamics
|
physiochemical or receptor interactions
|
|
|
|
reversible receptor bonds
|
ionic, Van der waals, Hydrogen
|
|
|
|
irreversible receptor bonds
|
covalent
|
|
|
|
EC50 (definition) (tells you what about the agonist?)
|
effective concentration for 50% response
|
relates to affinity of agonist for receptor
|
|
|
graded response graph (for drug effects)
|
linear format
|
|
|
|
dose-response graph (for drug effects)
|
logarithmic
|
|
|
|
threshold of dose-response graphs (definition) (gives you what info about the agonist?)
|
dose of agonist at which response begins
|
relates the affinity of agonist for the receptor
|
|
|
Effect = (of certain dose of drug)
|
E(max) [D] ---------------- K(D) + [D]
|
K(D) = EC50
|
|
|
intrinsic activity (definition) (influences?)
|
ability to stimulate a receptor once bound
|
efficacy and potency
|
|
|
E(max) (definition) (what % of receptor occupancy needed?)
|
maximum response
|
not all receptors need to be occupied
|
|
|
More efficacious agonists need ______ receptors occupied than less efficacious agonist to achieve the same effect.
|
less
|
|
|
|
Less efficacious agonists need ______ receptors occupied than more efficacious agonist to achieve the same effect.
|
more
|
|
|
|
secondary receptors
|
outside target tissue - mediate side-effects
|
|
|
|
strongest agonist has ______ intrinsic activity
|
strongest
|
|
|
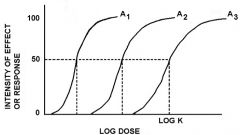
These agonists differ by their _____ but not in _____
(how can you tell?) |
binding, activating the receptor
|
different EC50's but same E(max)
|
|
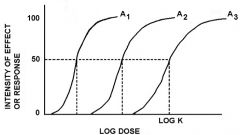
three agonists with the same?
|
intrinsic activity
|
|
|
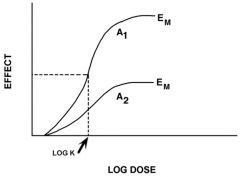
two agonists with different?
(but the same?) |
intrinsic activity
|
receptor binding affinity (K(D) is the same)
|
|
|
Efficacy
(depends on the?) |
the ability of the drug to activate the effector portion of the receptor once bound
|
structure of the drug
|
|
|
Potency
(depends on?) |
relates to the amount of drug that is needed for an effect
|
3)Biologic System (receptor density, efficiency of stimulus-response mech)
2)Interaction with the Receptor (affinity, efficacy) |
|
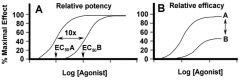
double-headed arrows represent differences in?
|
A- relative potency (amount of drug needed)
|
B- relative efficacy (maximum effect)
|
|
|
adding of a competative antagonist to a dose of agonist inc/dec the ____ of the Log Dose-response curve of the agonist?
|
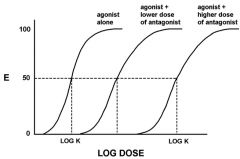
increases
|
K(D) or EC50
|
|
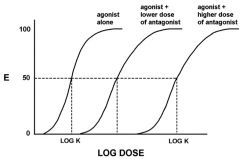
what type of antagonism?
|
non-competitive
|
|
|
|
(T/F) Non-competitive inhibitors inactivate the effector
|
(T)
|
|
|
|
(T/F) Noncompetitive antagonists interfere with agonist binding.
|
(T)
|
|
|
|
(T/F) Competitive antagonists interfere with agonist binding.
|
(F)
|
|
|
|
(T/F) Competitive inhibitors inactivate the effector.
|
(F)
|
|
|
|
characteristics of Simple Diffusion
|
Lipid Soluble, small, nonionized
|
|
|
|
Characteristics of Facilitated Diffusion
|
selective, can be saturated
|
|
|
|
Characteristics of Filtration
|
driven by hydrostatic pressure, transported through pores or channels between cells, size limiting
|
|
|
|
Amount of:
Intracellular Fluid Interstitial Fluid Plasma |
28L
9L 3L |
|
|
|
What characteristics do drug molecules have that can pass through the capillary membrane?
|
small molecule, lipid-(in)soluble
|
|
|
|
weak acids ____ a proton
|
give up
|
|
|
|
weak bases ___ a proton
|
accept
|
|
|
|
Henderson-Hasselbach Equation
|
pH= pKa + log A-/HA
|
|
|
|
which is greatly absorbed in the stomach, weak acids or weak bases?
|
weak acids
|
|
|
|
Two fluid compartments, 1: pH=7.4, 2: pH=1.4. Which direction will equilibrium shift for a weak acid?
|
Toward compartment 1.
|
|
|
|
Two fluid compartments, 1: pH=7.4, 2: pH=1.4. Which direction will equilibrium shift for a weak base?
|
Toward compartment 2
|
|
|
|
What is trapped in the stomach, weak acids or weak bases?
|
weak bases
|
|
|
|
Enteral drug administration.
|
GI tract
|
|
|
|
Parenteral Drug Administration.
|
any route of administration not through the GI tract.
|
|
|
|
Which is less painful and more rapid in reference to absorption, S.C. or intramuscular injections?
|
intramuscular
|
|
|
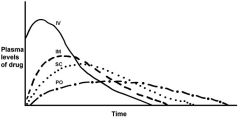
label routes of administration. Between which two is inhalation?
|
A-IV
B-IM C-SC D-PO and between A and B |
|
|
|
Bioavailability
(definition) (equation) |
fraction of dose available for biologic action
|
AUC (oral)
--------------- X100 AUC (iv) |
|
|
Equation to predict plasma concentration of drug- IV
(pill) |
Cp = Dose/Vd
(Vd = volume of distribution) |
Cp = (F x Dose)/Vd
(F = fraction absorbed) |
|
|
Volume of Distribution
(equation) (values) |
Vd = Dose / Plasma Concentration
(D/Cp) |
40L - sml molec. & lipid-soluble
12L- lipid-soluble 3L- protein bound |
|
|
Why might the Volume of Distribution of a drug appear to be more than 40?
|
Tissue Binding or plasma protein binding
|
|
|
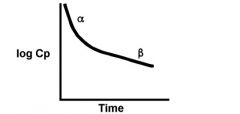
what are the two components of volume distribution of a drug?
|
a = drug distribution
b = drug elimination |
|
|
|
Factors affecting distribution of a drug:
|
Blood flow
Ability to enter fluid space (pH, binding, transport, solubility...) Time after administration Redistribution Size of Pt |
|
|
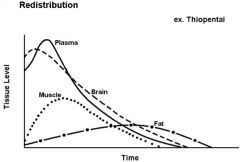
label initial compartments of drug distribution
|
Top- Plasma
Dashed- Brain Dotted- Muscle lowest- fat |
|
|
|
What do you do with an overdose of a weak acid drug for CNS toxicity? (aspirin/barbiturates)
|
increase plasma pH (with NaHCO3)
|
|
|
|
What do you do with an overdose of a weak base drug for CNS toxicity?
|
decrease pH (with HCl)
|
|
|
|
What can cause a smaller apparent Vd for a drug?
|
reporting of total measured drug in the plasma with drug plasma protein binding
|
|
|
|
loading dose
|
needing to fill the storage sites of a drug before enough free drug is available to interact with its target tissue... where are these storage sites?
|
short term- protein binding
long term- lipid or bone |
|
|
why is it dangerous to give both antibiotics and anticoagulants?
|
drug displacement- the antibiotic will displace some of anticoag attached to plasma proteins, releasing more free drug
|
|
|
|
Purpose of Biotransformations
|
to clear a drug from the plasma
|
|
|
|
Basics of preparing a drug for excretion
|
make it larger, charged, and more water soluble
|
|
|
|
What are the reactions that compose the first two Phases of biotransformation?
|
Phase 1: oxidation, reduction, hydrolysis
|
Phase 2: conjugation
|
|
|
term used to describe an increase in metabolism of the primary drug or other drugs in the Liver?
what is the consequence of this? |
induction
|
you may need to increase the dose of the drug to achieve the desired effect
|
|
|
term used to describe when a pt on antibiotics and cardiac drugs experiences an overdose of the cardiac drugs due to the antibiotics blocking the metabolism of the cardiac drugs
|
inhibition
|
|
|
|
(T/F) The hepatic microsomal drug metabolizing system performs both Phase 1 and Phase II processes
|
True
|
|
|
|
(T/F) The non-microsomal systems perform both Phase 1 and Phase II processes.
examples of non-microsomal systems? |
False- usually phase I (acetyl cholinesterase, alcohol dehydrogenase)
plasma, red cells |
|
|
|
What is the most common Phase 1 reaction for the biotransformation of drugs?
|
drug oxidation
|
|
|
|
What is the most common Phase 1 reaction for the biotransformation of drugs?
|
drug oxidation
|
|
|
|
Locations (organ and cellular specific) of oxidation of drugs
|
plasma, red cells, other tissues
ER, cytoplasm, mitochondria |
|
|
|
6 components of hepatic mixed function oxidase system
|
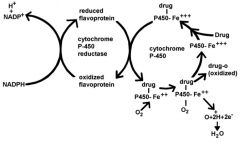
1) NADPH
2) cytochrome P450 reductase (flavoprotein) 3) cytochrome P450 (hemoproteins) 4) Mg++ 5) phospholipid 6) O2 |
|
|
|
In the hepatic mixed oxidase system does the drug bind to Ferrous or Ferric iron-P450?
|
Ferric +++ iron
|
|
|
|
Tylenol Overdoses deplete the body of NADPH. How does this effect the drug metabolism in the liver?
|
greatly reduces it. without NADPH cytochrome P-450 reductase oxidized flavoprotein can not be recycled to its reduced state, thus durg-P450-Fe+++ can not be reduced
|
|
|
|
(T/F) 1-2% of caucasians but 5-10% of SE asians have poor metabolic activity
|
False- %'s are reversed. The 4 types of phenotypes of metabolic activity are: poor, intermediate, extensive, and untrarapid
|
|
|
|
cellular areas where drug reductions take place in the liver
|
1) microsomes
2) cytoplasm 3) mitochondria |
|
|
|
(T/F) Phase 1 of drug biotransformation does not inactivate the drug
|
False- products of Phase 1 have variable activities, some active, some inactive
|
|
|
|
Location of Phase II biosynthetic reactions
|
Liver
|
|
|
|
The resulting molecule of a Phase II biotransformation reaction is:
|
larger, charged, water soluble, and inactive
|
|
|
|
What type of energy source is used for the Phase II conjugation reaction of glucaronide to a drug?
|
UTP
|
|
|
|
Which of the following are components of Phase I and which are Phase II biotransformation reactions?
1) acetyl Co-A 2) H20 3) Fe-+++ 4) S-adenosylmethionine 5) cytochrome P-450 6) UTP 7) Flavoprotein 8) H 9) Glucuronide |
1) acetyl Co-A: Phase II
2) H20: Phase I (hydrolysis) 3) Fe-+++: Phase I (oxidation) 4) S-adenosylmethionine (Phase II) 5) cytochrome P-450: Phase I (oxidation) 6) UTP: Phase II 7) Flavoprotein: Phase I (oxidation) 8) H: Phase I (reduction) 9) Glucuronide: Phase II |
|
|
|
equation for enzyme reaction with a drug
|
Vmax [D]
V=-------------- Km + [D] |
|
|
|
What order of kinetics occurs at low drug concentrations? (when [D],,Km)
|
First Order
|
|
|
|
What order of kinetics occurs when there is a large concentration of drug? ([D]>>Km)
|
Zero Order
|
|
|
|
Explain the enzyme/durg concentration relationship for zero order kinetics.
|
the enzyme is at its max efficiency
|
|
|
|
Explain the enzyme/durg concentration relationship for First order kinetics.
|
enzyme is proportionately responsive to the concentration of drug
|
|
|
|
(T/F) The hepatic microsomal drug metabolizing system experiences both induction and inhibition.
|
True
|
|
|
|
(T/F) The non-microsomal systems experience both induction and inhibition.
|
False- they only experience inhibition
|
|
|
|
what does saturation of the transport systems in the kidney due to the order of the kinetics of renal clearance?
|
changes First order to Zero order (max rate)
|
|
|
|
In manipulating the pH of urine (pH partitioning) to enhance the renal excretion of a drug, is the urine made to be more acidic or more alkaline than the plasma?
Is the plasma more acidic or alkaline than the brain? |
urine is more alkaline than the plasma (pH=8)
plasma is more alkaline than the brain (pH=7.5) Brain pH=7.4 |
|
|
|
What compound is used to alkalinize the urine?
1) Sodium Sulfate 2) Sodium Bicarbonate 3) Phenobarbital |
Sodium Bicarbonate
|
|
|
|
What is the role of activated charcoal in overdoses that do not involve the ingestion of the drug?
|
To stop the enterohepatic cycling that will keep the drug from being eliminated in bile.
|
|
|
|
equation for Clearance
|
Clearance Total =
C(metabolic) + C(renal) |
|
|
|
clearance rate of inulin =
|
filtration rate
|
|
|
|
clearance rate of glucose =
|
zero
|
|
|
|
clearance of PAH =
|
active transport + filtration (~650ml/min)
|
|