Reading...
![]()
Play button
![]()
Play button
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
95 Cards in this Set
- Front
- Back
|
What does hemolytic anemia result in? |
- Loss of RBC mass
- Release of cellular contents |
|
|
What are the inherited / intrinsic disorders that cause Hemolytic Anemia?
|
- RBC membrane disorders
- RBC enzyme deficiencies - Hemoglobinopathies |
|
|
What are the acquired / extrinsic disorders that cause Hemolytic Anemia?
|
- Auto-immune (Ab-mediated) hemolysis (warm and cold)
- Mechanical trauma - Infections (malaria) - Chemical - Splenic sequestration |
|
|
Where does pathologic hemolysis occur?
|
- Extravascular: most commonly, spleen >>> liver, BM
- Intravascular: less common, within the vessels - Both |
|
|
What happens to the RBCs that are not deformable?
|
- They are unable to squeeze through endothelium
- Get caught outside of sinusoid which is rich in macrophages - RBCs that can't get inside of the sinusoid get chewed up and engulfed by macrophage |
|
|
Which types of hemolytic anemias have RBCs hemolyzed in the extravascular space?
|
- RBC membrane deficiencies
- RBC enzyme deficiencies* - Sickle cell anemia* - Thalassemias - Auto-immune hemolysis (by antibodies) (* also intravascular hemolysis) |
|
|
Which types of hemolytic anemias have RBCs hemolyzed in the intravascular space?
|
- RBC enzyme deficiencies*
- Sickle cell anemia* - Auto-immune hemolysis (by complement) - PNH - Mechanical - Malaria (* also extravascular hemolysis) |
|
|
What are the morphologic characteristics in hemolytic anemia?
|
Normocytic anemia (normal in size)
|
|
|
What are the lab findings in hemolytic anemia?
|
- Reticulocytosis
- ↑ LDH, AST, and K+ (release of cellular contents) - Bilirubinemia - Hemoglobinemia - Hemoglobinuria - Hemosiderinuria |
|
|
What type of hemolysis has more prominent Hemoglobinemia? What happens to the free Hb?
|
- More pronounced in intravascular hemolysis
- Scavenged by Haptoglobin |
|
|
What are the types of Bilirubinemia?
|
- Unconjugated bilirubinemia (indirect)
- Conjugated bilirubinemia w/ liver disease |
|
|
What type of hemolysis has more prominent Hemoglobinuria?
|
- Mostly observed in intravascular processes
- Hb not scavenged by Haptoglobin |
|
|
What type of hemolysis has more prominent Hemosiderinuria? What happens?
|
- Mostly observed in intravascular processes
- Iron deposits in renal epithelium, then shed in urine |
|
|
How do lab tests assess the presence / concentration of Hb or heme in the blood or urine? What is the laboratory caveat with hemolysis?
|
Spectrophotometry using Beer's law:
A = ε * L * c - Absorbance is proportional to concentration - Spectrophotometer will absorb the red from the hemolyzed specimen, so it is less accurate - Hemoglobin absorbs at measured wavelengths, therefore, many assays produce falsely increased results in the setting of hemolysis - Could be a defect of poor phlebotomy (shearing the cells) or because they really had a hemolytic event |
|
|
What other lab test is a marker of hemolysis? How do you tell if it is a real hemolysis or false hemolysis?
|
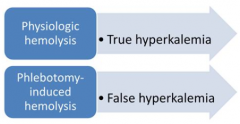
Hyperkalemia
- If you suspect that it is a phlebotomy-induced hemolysis you should do another lab draw to see if the same results occur or not |
|
|
What are the clinical features of hemolysis?
|
- Anemia
- Hyperbilirubinemia - Other characteristic symptoms are related to the underlying pathology causing the hemolysis (eg, vaso-occlusive crises in sickle cell anemia) |
|
|
What is a major determinant of the symptoms in hemolytic anemia?
|
Acuity of hemolysis:
- Most important question is how does patient's Hb and Hematocrit compare to baseline - Young patients w/ chronic hemolytic conditions (eg, sickle cell, thalassemia, pyruvate kinase deficiency) |
|
|
How low of a hemoglobin can a young patient with chronic hemolytic anemia have and still not have symptoms?
|
- Patients: 6-7 g/dL
- Normal: 12-16 g/dL |
|
|
What is the approximate hemoglobin conc. when there is <20% hemolysis? Symptoms?
|
> 11 g/dL
Restlessness |
|
|
What is the approximate hemoglobin conc. when there is 20-30% hemolysis? Symptoms?
|
- 10-11 g/dL
- Anxiety, dyspnea w/ exertion - Orthostatic hypotension - Tachycardia w/ exertion |
|
|
What is the approximate hemoglobin conc. when there is 30-40% hemolysis? Symptoms?
|
- 8-9 g/dL
- Syncope - Orthostatic hypotension - Tachycardia at rest |
|
|
What is the approximate hemoglobin conc. when there is >40% hemolysis? Symptoms?
|
- <8 g/dL
- Confusion - Shock |
|
|
What are the lab signs of hyperbilirubinemia in hemolytic anemia?
|
- Increased production of unconjugated (or indirect) bilirubin
- Direct bilirubin level is normal - Rare to have indirect bilirubin >5 mg/dL d/t hemolysis in absence of concurrent liver disease |
|
|
What are the clinical signs and when can you detect the clinical signs of hyperbilirubinemia in hemolytic anemia?
|
- Jaundice is detectable in most fair-skinned individuals when bilirbuin exceeds 2.5 mg/dL
- Scleral icterus can be detected by experienced physicians when bilirubin exceeds 1.5 mg/dL - Increased risk of bilirubin gallstones (pigmented) |
|
|
What are the rare complications of hemolytic anemia?
|
- Pigment induced kidney injury
- Folate deficiency d/t increased utilization - Increased risk of venous and arterial thrombosis related, including venous thrombosis of atypical sites (portal vein, cerebral venous sinus) |
|
|
What can cause kidney injury in hemolytic anemia?
|
- Heme pigment can cause tubular obstruction, direct cellular injury, and vasoconstriction resulting in decreased medullary blood flow
- Urinalysis shows hemoglobinuria w/o RBCs |
|
|
Which types of hemolytic anemia cause splenomegaly and hepatomegaly related to extra-medullary hematopoiesis?
|
- Thalassemia
- PK deficiency - Hereditary spherocytosis |
|
|
Which types of hemolytic anemia causes skeletal changes related to expansion of marrow, "chipmunk facies", convex bones?
|
β-thalassemia major
|
|
|
Which types of hemolytic anemia causes asplenia?
|
Sickle Cell Anemia
|
|
|
Which types of hemolytic anemia causes microangiopathic hemolytic anemia?
|
- TTP (Thrombotic thrombocytopenic Purpura)
- HUS (Hemolytic-Uremic Syndrome) - DIC (Disseminated Intravascular Coagulation) - Malignant HTN - CREST syndrome - Vasculitis - HELLP syndrome |
|
|
What does microangiopathic hemolytic anemia cause?
|
Microvascular infarction causes:
- Acute kidney injury - Liver disease - Abdominal pain - Fever - Mental status changes - Thrombocytopenia - Rash - Hemorrhagic diarrhea |
|
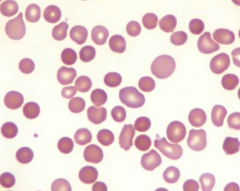
Case 1: 35 yo female presents to establish care. She is a poor historian and vaguely recalls a diagnosis of anemia as a child. She denies fatigue, headache, SOB, difficulty w/ exertion, palpitations, or abdominal discomfort. She has mild jaundice, splenomegaly, and laparoscopic surgical scars on abdomen. Hb is 9.4 g/dL. PB smear shows abnormal RBCs.
What RBC parameters are associated with this abnormal RBC morphology? |
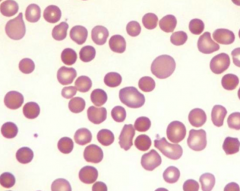
- Increased MCHC
- Increased RDW - Decreased MCV - Decreased RBC count (anemic) |
|
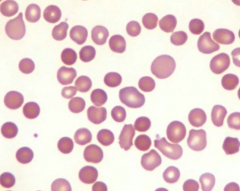
Case 1: 35 yo female presents to establish care. She is a poor historian and vaguely recalls a diagnosis of anemia as a child. She denies fatigue, headache, SOB, difficulty w/ exertion, palpitations, or abdominal discomfort. She has mild jaundice, splenomegaly, and laparoscopic surgical scars on abdomen. Hb is 9.4 g/dL. PB smear shows abnormal RBCs.
This RBC morphologic abnormality may be identified in what kind of conditions? |
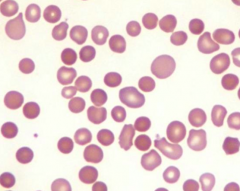
Acquired (auto-immune hemolytic anemia) and Inherited (hereditary spherocytosis)
|
|
|
Case 1: 35 yo female presents to establish care. She is a poor historian and vaguely recalls a diagnosis of anemia as a child. She denies fatigue, headache, SOB, difficulty w/ exertion, palpitations, or abdominal discomfort. She has mild jaundice, splenomegaly, and laparoscopic surgical scars on abdomen. Hb is 9.4 g/dL. PB smear shows abnormal RBCs.
What lab test is most important in differentiating the causes of this RBC morphologic abnormality? |
Direct Antiglobulin Test - distinguishes between inherited (eg, hereditary spherocytosis) and acquired (eg, auto-immune hemolytic anemia) causes
|
|
|
What test can identify spherocytes besides a PB smear?
|
Osmotic fragility / lysis test
- Spherocytes are more fragile in hypotonic solutions - This test tells you that there are spherocytes present |
|
|
What are the characteristics of spherocytes?
|
- Normal sized RBCs
- Increased Hb concentration - Less flexible, get trapped in spleen |
|
|
What disorders have spherocytes?
|
- Hereditary spherocytosis
- Auto-immune hemolytic anemia (AIHA): cold and warm |
|
|
Case 1: 35 yo female presents to establish care. She is a poor historian and vaguely recalls a diagnosis of anemia as a child. She denies fatigue, headache, SOB, difficulty w/ exertion, palpitations, or abdominal discomfort. She has mild jaundice, splenomegaly, and laparoscopic surgical scars on abdomen. Hb is 9.4 g/dL. PB smear shows abnormal RBCs.
What previous surgeries are the abdominal scars most likely secondary to? |
Cholecystectomy (or partial splenectomy)
|
|
|
Case 1: 35 yo female presents to establish care. She is a poor historian and vaguely recalls a diagnosis of anemia as a child. She denies fatigue, headache, SOB, difficulty w/ exertion, palpitations, or abdominal discomfort. She has mild jaundice, splenomegaly, and laparoscopic surgical scars on abdomen. Hb is 9.4 g/dL. PB smear shows abnormal RBCs.
The pathophysiology of this patient's disease is best characterized as? |
Congenital RBC membrane disorder
|
|
|
What are the congenital RBC membrane disorders?
|
- Hereditary spherocytosis
- Hereditary elliptocytosis |
|
|
What is the pathogenesis of hereditary spherocytosis?
|
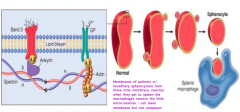
- Membranes of patients form little membrane vesicles
- When they get to spleen the macrophages remove the micro-vesicles - Cells lose membrane but not cytoplasm |
|
|
What is the most common inherited hemolytic anemia in the US? How is it inherited?
|
Hereditary Spherocytosis
- 1/5000 - 75% autosomal dominant - 25% sporadic (recessive and new mutations, compound heterozygosity) |
|
|
What are the primary defects causing hereditary spherocytosis?
|
- Ankyrin (most common)
- Spectrin - Band 3 - Most patients have deficiencies in both ankyrin AND spectrin d/t a protein imbalance |
|
|
What are the clinical findings of hereditary spherocytosis?
|
- Wide variability in severity of hemolysis
- Sometimes an incidental finding - Mild anemia - Moderate to severe hemolysis - Splenomegaly |
|
|
How do you treat hereditary spherocytosis?
|
- Depends on clinical severity
- Manage chronic hemolysis (may require transfusions or splenectomy) - Treat gallstones and jaundice w/ cholecystectomy (usually by age 25-30) - Managing aplastic crisis (mostly by treating Parvovirus B19) - Splenectomy (complete for adults and partial for children under age 6) |
|
|
At what age is it safe to do a full splenectomy vs partial splenectomy? Why?
|
- Before age 6 do a partial splenectomy
- Full splenectomy is discouraged in children <6 because of increased incidence of encapsulated organism infections (eg, strep pneumo) - But the partial splenectomy is still helpful because it decreases need for transfusions |
|
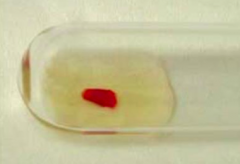
Case 1b: Patient with similar findings and laboratories presents to ED w/ new onset jaundice, fatigue, and shortness of breath. BP 142/88, P 100bpm, R 18, T 98.6, and SaO2 97%. Bilirubin is 3 mg/dL (normal 0.2-1.2 mg/dL). They have the same PB smear as previous patient.
Results of DAT are shown. What is the most likely cause? |
Classically a young woman with lupus (auto-immune disorder) causing Auto-Immune Hemolytic Anemia
|
|
|
Case 1b: Patient with similar findings and laboratories presents to ED w/ new onset jaundice, fatigue, and shortness of breath. BP 142/88, P 100bpm, R 18, T 98.6, and SaO2 97%. Bilirubin is 3 mg/dL (normal 0.2-1.2 mg/dL). They have the same PB smear as previous patient.
What is the most appropriate therapeutic option for this patient? |
Corticosteroids to treat Auto-Immune Hemolytic Anemia
|
|
|
What is the pathophysiology responsible for Auto-Immune Hemolytic Anemias (AIHAs)?
|
- Host antibodies reactive w/ autologous RBCs
- Shortened in vivo RBC survival |
|
|
How do you distinguish the different types of Auto-Immune Hemolytic Anemias (AIHAs)?
|
- Warm auto-Abs → 37°C optimal temperature of activity
- Cold auto-Abs → 0-5°C optimal temperature range of activity |
|
|
Which type of Auto-Immune Hemolytic Anemia (AIHA) is more common? What are the causes?
|
Warm auto-antibody type (~80%):
- Idiopathic (primary) - Secondary |
|
|
What disorders are associated with secondary Warm Auto-Immune Hemolytic Anemias (AIHAs)?
|
- Lymphoproliferative disorders (excessive production of Abs against RBCs)
- Auto-immune disorders (eg, Lupus) - Non-hematopoietic neoplasms (eg, ovarian cancer) - Drugs (eg, α-methlydopa, cephalosporins) |
|
|
Which drugs are associated with secondary Warm Auto-Immune Hemolytic Anemias (AIHAs)?
|
- α-methyldopa
- Cephalosporins |
|
|
Which type of Auto-Immune Hemolytic Anemia (AIHA) is less common? What are the causes?
|
Cold auto-antibody type (~20%):
- Idiopathic (primary) - Secondary |
|
|
What disorders are associated with secondary Cold Auto-Immune Hemolytic Anemias (AIHAs)?
|
- Post-infectious (eg, infections mononucleosis, Mycoplasma)
- Lymphoproliferative disorders (excessive production of Abs against RBCs) |
|
|
What is the pathogenesis of Warm Auto-Immune Hemolytic Anemias (AIHAs)?
|
Extravascular Hemolysis
- Ab mediated (IgG1 and IgG3) OR - Complement mediated (classical pathway) - Leads to Spherocyte production |
|
|
What is the pathogenesis of Cold Auto-Immune Hemolytic Anemias (AIHAs)?
|
- IgM binds in cold
- IgM-mediated agglutination - RBC agglutination impedes blood flow in superficial distal vessels - IgM and complement mediated RBC injury (extravascular hemolysis in liver > spleen) |
|
|
What are the symptoms of Warm Auto-Immune Hemolytic Anemias (AIHAs)?
|
- Symptoms of anemia
- Mild → severe (w/ massive hemolysis) - Organomegaly |
|
|
What are the symptoms of Cold Auto-Immune Hemolytic Anemias (AIHAs)?
|
Acute or chronic form
- Acute form occurs 2-3 weeks into infectious illness - Follows cold exposure Symptoms of anemia - Mild → severe (w/ massive hemolysis) Acrocyanosis (bluish/purplish skin) - Raynaud phenomenon |
|
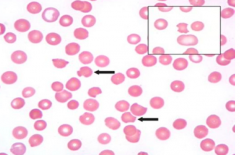
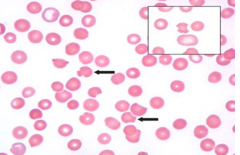
Case 2: 17 yo AA male presents w/ persistent fatigue following recent sinus infection. Patient was started on trimethoprim-sulfamethaxazole 7 days ago and has since had resolution of fevers. Physical exam reveals scleral icterus. Hb 8.1 g/dL. MCV 105 fL. Absolute reticulocyte count 185,000 / µL (normal 30,000-82,000/µL). Bilirubin 7.3 mg/dL (normal <1.2). Direct bilirubin normal.
What genetic mutation most likely explains this patient's symptoms and lab findings? |
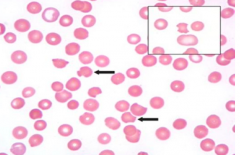
Glucose 6-Phosphate Dehydrogenase
|
|
Case 2: 17 yo AA male presents w/ persistent fatigue following recent sinus infection. Patient was started on trimethoprim-sulfamethaxazole 7 days ago and has since had resolution of fevers. Physical exam reveals scleral icterus. Hb 8.1 g/dL. MCV 105 fL. Absolute reticulocyte count 185,000 / µL (normal 30,000-82,000/µL). Bilirubin 7.3 mg/dL (normal <1.2). Direct bilirubin normal.
RBC abnormality observed in PB smear is secondary to what? |
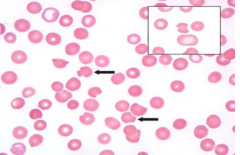
Mononuclear cell phagocytosis of denatured protein (Heinz bodies made of denatured Hb d/t deficiency of G6PD)
|
|
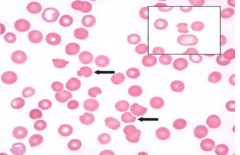
Case 2: 17 yo AA male presents w/ persistent fatigue following recent sinus infection. Patient was started on trimethoprim-sulfamethaxazole 7 days ago and has since had resolution of fevers. Physical exam reveals scleral icterus. Hb 8.1 g/dL. MCV 105 fL. Absolute reticulocyte count 185,000 / µL (normal 30,000-82,000/µL). Bilirubin 7.3 mg/dL (normal <1.2). Direct bilirubin normal.
Based on the lab findings, what is true about the patient's BM? |
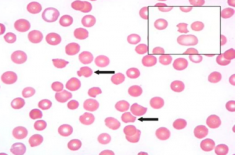
- Erythroid precursors outnumber the granulocytic precursors
- This is true of all hemolytic anemias because they will rev up production of erythroid precursors to compensate for those that are being hemolysed |
|
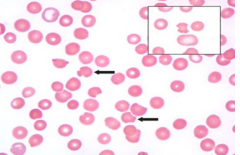
Case 2: 17 yo AA male presents w/ persistent fatigue following recent sinus infection. Patient was started on trimethoprim-sulfamethaxazole 7 days ago and has since had resolution of fevers. Physical exam reveals scleral icterus. Hb 8.1 g/dL. MCV 105 fL. Absolute reticulocyte count 185,000 / µL (normal 30,000-82,000/µL). Bilirubin 7.3 mg/dL (normal <1.2). Direct bilirubin normal.
What is the most appropriate treatment at this time? |
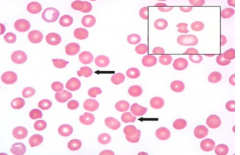
Stop trimethoprim-sulfamethaxaole (this is causing the oxidative stress that his G6PD deficiency can't keep up with → Heinz bodies → macrophages make bite cells)
|
|
|
Case 2: 17 yo AA male presents w/ persistent fatigue following recent sinus infection. Patient was started on trimethoprim-sulfamethaxazole 7 days ago and has since had resolution of fevers. Physical exam reveals scleral icterus. Hb 8.1 g/dL. MCV 105 fL. Absolute reticulocyte count 185,000 / µL (normal 30,000-82,000/µL). Bilirubin 7.3 mg/dL (normal <1.2). Direct bilirubin normal.
What kind of disorder is this? |
Congenital RBC enzyme disorder (G6PD)
|
|
|
Case 2: 17 yo AA male presents w/ persistent fatigue following recent sinus infection. Patient was started on trimethoprim-sulfamethaxazole 7 days ago and has since had resolution of fevers. Physical exam reveals scleral icterus. Hb 8.1 g/dL. MCV 105 fL. Absolute reticulocyte count 185,000 / µL (normal 30,000-82,000/µL). Bilirubin 7.3 mg/dL (normal <1.2). Direct bilirubin normal.
What should you counsel the patient about? |
Similar episodes with overwhelming infections
- They have a tendency to hemolyze if they are in the presence of something that stimulates oxidative stress - Normal machinery (G6PD) can deal w/ oxidant stress but those that have deficiency are susceptible to insults |
|
|
What pathways can glucose go down?
|
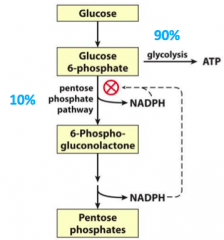
- 90% for Glycolysis
- 10% for Pentose Phosphate Pathway |
|
|
What regulates the partitioning into glycolysis vs the pentose phosphate pathway?
|
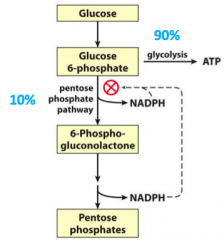
NADPH
|
|
|
What pathway does a deficiency of Glucose 6-Phosphate Dehydrogenase affect?
|
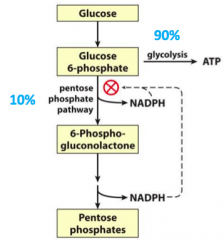
Pentose Phosphate Pathway
|
|
|
What is the congenital RBC enzyme disorder to know? Implications?
|
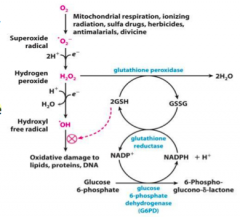
Glucose 6-Phosphate Dehydrogenase Deficiency
- Defect in the pathway responsible for reducing oxidants - Causes a build up of Glucose 6-Phosphate - Decreased glutathione production - Inability to reduce oxidant stressors |
|
|
What are some oxidant stressors that can affect a patient with Glucose 6-Phosphate Dehydrogenase Deficiency?
|
- Infections
- Fava benas - Drugs: dapsone, sulfonamides, primaquine, nitrofurantoin, and quinolones |
|
|
What happens to RBCs in Glucose 6-Phosphate Dehydrogenase Deficiency?
|
- Increased susceptibility to oxidant stress
- RBCs lead to hemolytic anemia (extravascular process) |
|
|
How common is Glucose 6-Phosphate Dehydrogenase Deficiency? How is it obtained?
|
- 400 million affected
- Common in malarial endemic areas - X-linked disorder |
|
|
Which Glucose 6-Phosphate Dehydrogenase Deficiency variant is most common in USA? Implications?
|
G6PD A-
- Moderate hemolysis - Found in 11% of AAs G6PD A and B are both normal and do not cause hemolysis |
|
|
How does a Glucose 6-Phosphate Dehydrogenase Deficiency affect males?
|
X-linked
- Normal male will have 100% G6PD activity - Deficient male will have 5-15% GDPD activity as RBC ages |
|
|
How does a Glucose 6-Phosphate Dehydrogenase Deficiency affect females?
|
X-linked
- Normal female will have 100% G6PD activity - Female carrier will have ~50% G6PD activity because X inactivation leads to 2 separate RBC populations |
|
|
What is the hematologic impact of dapsone (oxidant stressor) on a normal person?
|
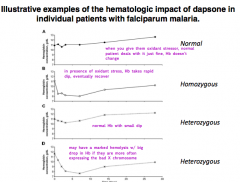
When you give them oxidant stressor, normal patient deals with it just find, Hb doesn't change
|
|
|
What is the hematologic impact of dapsone (oxidant stressor) on a homozygous patient (male w/ G6PD deficiency)?
|
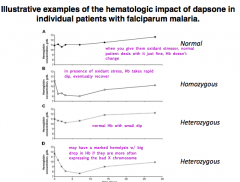
Oxidant stress causes Hb to rapidly dip, eventually they recover
|
|
|
What is the hematologic impact of dapsone (oxidant stressor) on a heterozygous female with 50% activity?
|
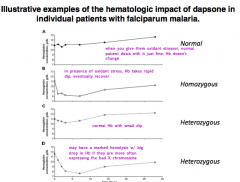
Normal Hb with small dip
|
|
|
What is the hematologic impact of dapsone (oxidant stressor) on a heterozygous female with greater expression of the X chromosome with low G6PD activity?
|
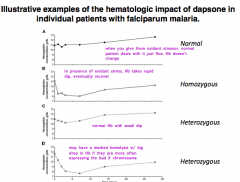
May have marked hemolysis w/ big drop in Hb
|
|
|
What are the features of G6PD A-?
|
- 11% of African-American males
- Leads to an unstable enzyme (100% activity in new RBCs but only 5-15% activity in old RBCS) - Self-limited hemolysis (old RBCs hemolyze, reticulocytes and new RBCs do not hemolyze) |
|
|
How do we diagnose Glucose 6-Phosphate Dehydrogenase Deficiency?
|
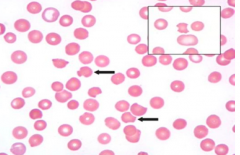
PB smear shows bite cells
|
|

Case 3: 42 yo AA male presents w/ headaches and altered mental status. PMH shows end-stage kidney disease secondary to long-standing HTN. BP 220/150 mmHg. Urinalysis shows 4+ proteinuria, +hemoglobinuria, but no RBCs. Elevated creatinine. PB smear shows schistocytes / fragments.
What lab abnormality is likely to be also present in this patient? |

Decreased haptoglobin
|
|
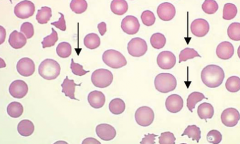
Case 3: 42 yo AA male presents w/ headaches and altered mental status. PMH shows end-stage kidney disease secondary to long-standing HTN. BP 220/150 mmHg. Urinalysis shows 4+ proteinuria, +hemoglobinuria, but no RBCs. Elevated creatinine.
What does the PB smear reveal (ignore arrows) about the BM function in this patient? |
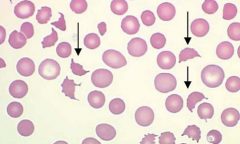
Erythroid compartment is responsive to a hemolytic insult (presence of reticulocytes)
|
|
|
Case 3: 42 yo AA male presents w/ headaches and altered mental status. PMH shows end-stage kidney disease secondary to long-standing HTN. BP 220/150 mmHg. Urinalysis shows 4+ proteinuria, +hemoglobinuria, but no RBCs. Elevated creatinine. PB smear shows schistocytes / fragments and reticulocytes.
The most likely diagnosis to explain the patient's symptoms and lab findings are? |
Malignant HTN
|
|

Case 3: 42 yo AA male presents w/ headaches and altered mental status. PMH shows end-stage kidney disease secondary to long-standing HTN. BP 220/150 mmHg. Urinalysis shows 4+ proteinuria, +hemoglobinuria, but no RBCs. Elevated creatinine. PB smear shows schistocytes / fragments and reticulocytes.
All of these patients may present with similar PB findings, except? |

5 yo w/ splenic sequestration would not present with schistocytes and reticulocytes
|
|
|
Case 3: 42 yo AA male presents w/ headaches and altered mental status. PMH shows end-stage kidney disease secondary to long-standing HTN. BP 220/150 mmHg. Urinalysis shows 4+ proteinuria, +hemoglobinuria, but no RBCs. Elevated creatinine. PB smear shows schistocytes / fragments.
The pathophysiology of this patient's disease is best characterized as? |
RBC fragmentation disorder
|
|
|
What can cause mechanical trauma to RBCs?
|
- Extreme heat / burn
- Defective cardiac valves - Microangiopathic hemolytic anemias |
|
|
What are the disorders that can cause Microangiopathic Hemolytic Anemias?
|
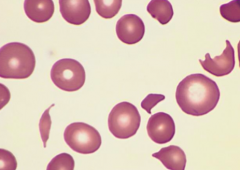
- Thrombotic Thrombocytopenic Purpura (TTP)
- Malignant HTN - Anti-phospholipid Ab Syndrome - Disseminated Intravascular Coagulation (DIC) - Disseminated cancer |
|
|
What happens to the vessels in malignant HTN? Implications for RBCs?
|
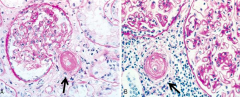
Hyaline Atherosclerosis - shears RBCs d/t thickening
|
|
|
What happens to the vessels in Thrombotic Thrombocytopenic Purpura? Implications for RBCs?
|
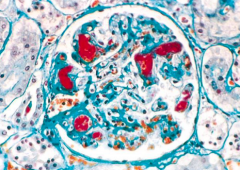
- Fibrin clots occlude RBC transport
- Leads to shearing of RBCs into fragments |
|
|
What disorder is caused by an acquired mutation in the PIGA gene?
|
Paroxysmal Nocturnal Hemoglobinuria (PNH)
|
|
|
What happens in Paroxysmal Nocturnal Hemoglobinuria (PNH)? Cause?
|
- Rare disorder
- Acquired mutation in PIGA gene on X chromosome - Results in decreased GPI-linked proteins |
|
|
What are the implications of the PIGA gene mutation in Paroxysmal Nocturnal Hemoglobinuria (PNH)?
|
- Lack of GPI-linked anti-complement proteins makes cells susceptible to complement lysis
- All myeloid lineages are affected, though RBCs are most sensitive to complement lysis |
|
|
What is the clinical presentation of Paroxysmal Nocturnal Hemoglobinuria (PNH)?
|
- Chronic low level hemolysis
- Acute, episodic hemolysis events - Hemoglobinuria - Venous thromboses |
|
|
How do you diagnose Paroxysmal Nocturnal Hemoglobinuria (PNH)?
|
Flow Cytometry for GPI-linked proteins
PNH if there is a deficiency of: - CD55 (DAF) - CD59 - Examined on WBCs and RBCs |