![]()
![]()
![]()
Use LEFT and RIGHT arrow keys to navigate between flashcards;
Use UP and DOWN arrow keys to flip the card;
H to show hint;
A reads text to speech;
113 Cards in this Set
- Front
- Back
|
Agglutination |
1) Clumping of red cells due to antibody coating 2) Main reaction we look for in blood banking |
|
|
Two stages of Agglutination |
1) Coating of cells ("sensitization") -Substances like LISS and PEG help overcome physical barriers to allow antigens and antibodies get closer to each other 2) Formation of bridges -Lattice-like structure formation -IgG isn't good at this; IgM antibodies are better both at bridge formation and fixing complement = hemolysis
|
|
|
Tube Testing 1) Immediate spin "phase" 2) 37 degree C "phase" 3) Indirect antiglobulin "phase" |
1) Serum + 2-5% RBC together in tube, centrifuge and examine 2) Above mixture + incubate, then centrifuge and examine -Often use potentiator (LISS/PEG) 3) Wash above mixture to remove UNBOUND globulins. Then add antihuman globulin, centrifuge and examine |
|
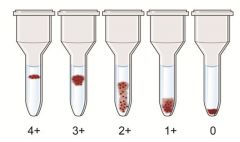
Gel Testing (Column Agglutination) |
1) Multiple microtubules filled with gel + anti-IgG reagent 2) Gel particles separate red cell clusters by size 3) Anti-IgG grabs onto red cells coated by IgG 4) Bigger red cell agglutinates will be stuck higher in gel 5) Negative gel tests show cells in a button at the bottom while positive tests have cells spread in varying degrees through the microtube |
|
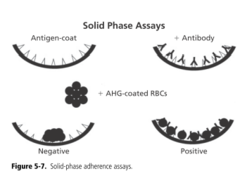
Solid-Phase Red Cell Adherence Testing |
1) Uses binding of Ab to RBCs that are themselves bound to the sides of microwells (manufacturer-created with Ag we are interested in) 2) Lab adds pt serum, incubates, washes; Ab if present binds to test RBCs 3) Indicator RBCs (coated with monoclonal anti-IgG) attach to test RBCs via bound Ab 4) Negative results - cells in a button at bottom because indicator cells don't bind to the test RBCs - no serum Ab in pt 5) Positive results - cells spread in a "carpet" because indicator cells have bound to test RBCs all along via the bound pt serum Ab |
|
|
Direct Antiglobulin Test ("Coomb's Test")
|
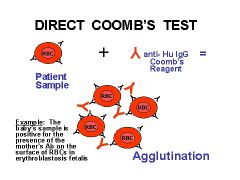
Red cells taken directly from pt, washed, then mixed with AHG; checks for in-vivo coating of RBCs with Ab and/or complement |
|
|
Indirect Antiglobulin Test (IAT) |
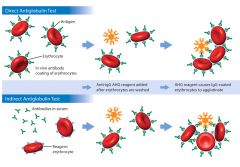
Indirect - checks for in-vitro coating of RBCs with Ab or complement |
|
|
IAT Variation |
1) Can be used to check for an unknown Ab by using RBCs with a known Ag profile - Ab screen 2) Can be used to check for an unknown red cell Ag by using serum with a known Ab specificity - RBC Ag testing 3) Can be used to check for a reacting known Ag and unknown Ab - crossmatch procedure |
|
|
Coomb's Control |
1) Used after negative DAT or IAT to ensure proper functioning of AHG reagent 2) IgG-coated RBCs added to AHG-cell mixture 3) Agglutination should occur, if negative = bad AHG or no AHG added |
|
|
Blood Groups showing Dosage |
Kidd Duffy Rh MNS
1) Certain Abs do not react as strongly with RBCs that have Ag coded for by a single gene (heterozygous) |
|
|
Neutralization ABO Lewis P1 Sda Chido, Rodgers |
ABO = Saliva (secretor) Lewis = Saliva (secretor for Leb) P1 = hydatid cyst fluid, pigeon egg fluid Sda = Urine Chido, Rodgers = Serum |
|
|
Significant vs. Insignificant Blood Group Antibody Characteristics |
Significant = tend to be "warm-reacting," and IgG; require exposure; cause HDN & HTRs
Insignificant = tend to be "cold-reacting," and IgM (Ice creaM); tend to be naturally occuring, no HDN* & no HTRs*
*Note that the ABO blood group is the one BIG exception (IgM and cold-reacting) |
|
|
Enzyme-Enhanced, Enzyme-Decreased, & Enzyme Unaffected |
Enhanced = ABO, Lewis, I/i, P, Rh, & Kidd Decreased = Duffy (Duffy destroyed) & MNS Unaffected = Kell |
|
|
ABO Blood Group Type I and Type II chains Se gene (FUT2) H gene (FUT1) |
1) Type I: Primarily glycoproteins in secretions and plasma carrying free-floating Ags 2) Type II: Primarily glycosphingolipids carrying bound Ags on RBCs 3) Se gene (FUT2): chromosome 19, 80% of us have this gene; a secretor is a person able to make A or B Ag in their secretions -Codes for a FUT enzyme that adds fucose to Type I chains = product is type 1 H Ag 4) H gene (FUT1): closely linked to Se on chromosome 19 -Codes for a FUT enzyme that adds fucose to Type II chains = product is type II H Ag -Virtually 100% frequency = lack is "Bombay phenotype" -H is required before A and/or B Ag can be made |
|
|
Group A sugar Group B sugar
Relative amounts of H by blood group |
Group A sugar = N-acetylgalactosamine Group B sugar = Galactose
H by blood group O > A2 > B > A2B > A1 > A1B |
|
|
Lectins and their specificity Dolichos biflorus Ulex europaeus Vicea graminea Arachus hypogea Glycium max Saliva |
Dolichos biflorus - A1, Sda Ulex europaeus - H Vicea graminea - N Arachus hypogea - T Glycium max - T, Tn Saliva - Tn |
|
|
Blood Group O |
1) Generally the most common 2) Genotype: OO 3) Antigen: H, no sugars transferred 4) Abs: anti-A, anti-B, anti-A,B -Characteristically IgM Ab and react strongly at body temp 5) Abs in group O people may have strong IgG component = may cross placenta to cause mild HDFN (most common cause of HDFN) |
|
|
Blood Group A |
1) Genotypes: AA, AO 2) Ag: A, H 3) Abs: anti-B (primarily IgM) 4) Subgroups: -A1 (80%) and A2 (~20%) -A1 RBCs have ~5x more A Ag than A2 cells -Small % of A2's form anti-A1 = can cause ABO discrepancies |
|
|
Blood Group B |
1) Genotypes: BB, BO 2) Ag: B, H 3) Abs: anti-A (primarily IgM) 4) B subgroups, usually unimportant and less frequent |
|
|
Blood Group AB |
1) Least frequent (~4%) 2) Ag: A & B (very little H) 3) Abs: None |
|
|
Acquired B phenotype |
1) Cause of unexpected Ag on ABO testing 2) Seen in contact with enteric gram negatives = colon cancer, intestinal obstruction, gram-negative sepsis 3) AB forward typing with weak rxn with reagent anti-B, types as A in reverse typing 4) Bacteria deacetylate group A sugar (GalNAc); remaining galactosamine (not galactose, but close enough) cross-reacts with reagent anti-B 5) To confirm = acidify serum (no reaction with anti-B), add acetic anhydride (re-acetylates), autoincubation (no reaction), BS-1 lectin (no reaction) |
|
|
Bombay Phenotype |
1) Total lack of H, A & B Ags due to lack of H and Se genes 2) Naturally occurring strong anti-h, anti-A, anti-B 3) Tests as O forward and reverse, but Ab screen wildly positive 4) Requires other Bombay donors |
|
|
Para-Bombay Phenotype |
1) Similar, but these pts have Se gene to partially compensate for lack of H 2) RBCs may type like Bombays, but serum/secretion testing shows free H and A or B Ag (unless group O) 3) Have anti-H in serum |
|
|
Consequences of ABO incompatibility |
Severe acute HTRs - most frequent cause of blood bank fatalities -> usually due to clerical errors Most frequent cause of HDFN; usually mild, however |
|
|
Lewis Blood Group |
1) Type I chains only (glycoprotein) 2) One gene = Le (FUT3) - adds fucose to subterminal GlcNAc which makes Le a 3) In a non-secretor, Le a is the only Lewis ag possible 4) Le a CANNOT be further modified to make Le b 5) In secretors, Se product adds fucose, then Le product adds fucose = this makes Le b -This occurs preferentially in secretors -Le b better at adsorbing onto the surface of RBCs |
|
|
Lewis phenotypes, Ags, & Abs |
1) Phenotypes: Le (a+b-), Le (a-b+), Le(a-b-) 2) 22% of blacks are Le(a-b-) vs. only 6% of whites 3) Abs are naturally occurring -Primarily in Le (a-b-) -Cold-reacting IgM -Neutralize with saliva 4) Rare HTRs (more commonly anti-Le a) 5) No HDFN = Ab doesn't cross placenta and Le Ag are not present on fetal RBCs |
|
|
Weird Stuff about Lewis |
1) Lewis Ags decrease during pregnancy -May appear Le (a-b-) and have transient, insignificant Lewis Abs -Thought to be due to increased plasma volume diluting the Ag 2) Le (a-b+) don't make anti-Le a 3) H. pylori may attach via Le b Ag 4) Le (a-b-) children have increased risk of UTI (E.coli) |
|
|
I/i Blood Group |
1) Expression is age-dependent -Simple chains in neonates make i Ag -More branched chains in adults make I Ag 2) Abs are cold-reacting IgM, naturally occurring, and auto-Abs are very common 3) Classic associations -Auto-anti-I = cold agglutinin disease, Mycoplasma pneumoniae infection -Auto-anti-i = associated with infectious mononucleosis |
|
|
P Blood Group |
1) P1 only P group Ag 2) Combination of P1, Pk, and P determine P phenotype 3) Very rare people lack all three and make anti-PP1Pk = associated with AHTRs, HDFN, and early, spontaneous abortions 4) P ag is parvovirus B19 receptor 5) Pk ag is receptor for various bacteria and toxins |
|
|
P Blood Group continued |
1) Antibodies are cold-reacting, naturally occurring, insignificant IgM 2) Titers elevated in those with hydatid cyst disease (Echinococcus) and bird handlers 3) Association with PNH (P for PNH) -Biphasic IgG with anti-P specificity -Binds in cold temps and hemolyzes when warmed -Donath-Landsteiner 4) Classically associated with syphilis, now with viral infections in children |
|
|
Rh Blood Group Nomenclature R1 R2 R0 Rz |
R1 = DCe R2 = DcE R0 = Dce Rz = DCE |
|
|
Rh Blood Group Nomenclature r' r" r ry |
r' = dCe r" = dcE r = dce ry = dCE |
|
|
Rh frequencies by race |
Whites = R1 > r > R2 > R0 Blacks = R0 > r > R1 > R2
R0 is most common in blacks, least common in whites r is always 2nd in frequency R1 always comes before R2 |
|
|
Rh Antibodies |
1) Exposure required 2) Warm-reacting IgG |
|
|
Consequences of Rh incompatibility |
1) Very immunogenic, D induces most Abs, then c and E. 2) Up to 80% of D- pts make anti-D with one unit D+ RBCs 3) Exposed = HTRs with extravascular hemolysis 4) Most severe and prototypical HDN |
|
|
Weak D phenotype |
1) Quantitative defect in D ag Causes: -"C in trans to D" -Weak form of RHD gene -Mosaic forms of D antigen; may lack portions of D antigen |
|
|
D-negative phenotype |
1) Unusual b/c caused by mutations and deletions 2) Caucasians - have del of Rh D gene 3) Blacks - point mutation in Rh D gene 4) Asians - usually have inactive Rh D gene |
|
|
Partial D phenotype |
1) Qualitative defect, but some have both quant and qual defect 2) Abs form against parts of RHD; Ab appears to be anti-D at first glance 3) Classic scenario: Anti-D in a D+ pt 4) Partial D moms need RHIg while weak D mom do not 5) Partial D recipients may make anti-D when receiving D+ RBCs, weak D recipients generally don't |
|
|
Rh null phenotype |
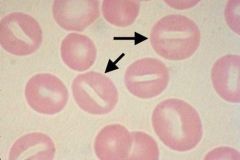
1) No Rh antigens at all 2) Hemolytic anemia with stomatocytes |
|
|
Kidd Blood Group |
1) Enzyme-enhanced 2) Jka, Jkb - b more common than a (reversed from most) 3) Exposure required 4) Warm-reacting IgG 5) Very good at fixing complement 6) Marked dosage effect 7) Variable ab expression - often disappears with time/storage |
|
|
Weird Stuff about Kidd |
1) Often the culprit of a delayed HTR 2) Anamnestic response 3) Intravascular hemolysis often severe 4) Mild HDFN at worst (child can only be one ag different than mom - remember dosage) |
|
|
MNS Blood Group |
1) Glycophorin A carries M & N antigens 2) Glycophorin B carries S, s, & U antigens 3) Frequency: - M = N - s > S 4) Enzymes destroy M, N & S but do not greatly affect s antigens |
|
|
anti-M/anti-N vs. anti-S/anti-s |
Anti-M & Anti-N: 1) Naturally occurring 2) Cold IgM 3) Show dosage and are insignificant Anti-S, Anti-s, and Anti-U: 1) Require exposure 2) Warm IgG 3) Show minimal dosage and are significant |
|
|
In 2% of African Americans see S-s- |
May also be U negative => lack glycophorin B |
|
|
N-like Antigen ('N') |
1) Glycophorin B has sequence that matched glycophorin A's last 5 - not actual N antigen, but close enough - ('N') 2) Keeps most M+N- people from making anti-N |
|
|
What antibody can be induced by hemodialysis? |
Anti-N 1) Formaldehyde sterilization of machine 2) Modification of the N antigen |
|
|
Duffy Blood Group |
1) Enzyme decreased 2) Fy b > Fy a (antigens) 3) Fy (a-b-) is most common Duffy phenotype in blacks (68%) - almost never in whites 4) Fy a much more common in Asians than in whites |
|
|
Duffy Antibodies |
1) Anti-Fy a much more common and significant 2) Require exposure 3) Warm-reacting IgG 4) Marked dosage effect 5) May have variable expression like Kidd
|
|
|
Consequences of Duffy incompatibility |
1) Severe HTRs, usually delayed and extravascular 2) Mild HDFN (for dosage reasons like Kidd) |
|
|
Weird stuff about Duffy |
1) Fy(a-b-) => malarial resistance to vivax and knowlesi |
|
|
Kell Blood Group |
Antigens: 1) Low frequency, K: 9% in whites, 2% in blacks), Js a, Kp a 2) High frequency, k: 99.8%, Js b, Kp b 3) Antigens destroyed by thiol reagents (2-ME, DTT, ZZAP) but not enzymes alone |
|
|
Kell Null phenotype |
1) Absence of all Kell antigens 2) Kx increased - may stabilize RBC membrane 3) Significant anti-Ku (universal) with exposure |
|
|
Kell Antibodies |
1) Anti-K: very common (most common non-ABO after anti-D) -Warm-reacting IgG 2) Anti-k: very uncommon due to high frequency -Warm-reacting IgG 3) Consequences: -Severe HTRs; may be acute or delayed, usually extravascular -Severe HDN |
|
|
McLeod Phenotype |
1) Absence of Kx 2) Markedly decreased (not absent) Kell system antigens 3) No anti-Ku 4) Hemolytic anemia with acanthocytes 5) Associated with CGD (X-linked) - susceptible to catalase-positive organisms (staph) 6) Associated with cardiac and nervous system abnormalities |
|
|
Lutheran Antigens |
1) Lu a and Lu b 2) Linked to Se on chromosome 19 3) Antibodies uncommon and not usually significant 4) Enzymes decrease Lu antigen activity |
|
|
HTLA |
1) High-titer, low-avidity 2) High frequency 3) Chido, Rodgers most frequent -Complement components (C4) 4) Clinically insignificant 5) Neutralize with serum |
|
|
Anticoagulant/Preservative Solutions |
Citrate-phosphate-dextrose (CPD) & Citrate-phosphate-dextrose-dextrose (CP2D) = 21 days RBC/whole blood storage
Citrate-phosphate-dextrose-adenine (CPDA-1) = 35 days of RBC/whole blood storage
Adenine Saline (AS-1, 3, 5) = increases shelf life of RBCs to 42 days (added into CPD) |
|
|
Product = PRBCs/whole blood
Storage = ? |
35 days (CPDA-1) 42 days (Additives) 1-6 C |
|
|
Product = Frozen RBCs
Storage = ? |
10 years; -65 C 24 hours after thawing |
|
|
Product = Washed RBCs
Storage = ? |
24 hours 1-6 C |
|
|
Product = Platelets
Storage = ? |
5 days 20-24 C Gentle agitation |
|
|
Product = WBCs
Storage = ? |
24 hours 20-24 C No agitation |
|
|
Product = FFP
Storage = ? |
1 year; -18 C OR 7 years; -65 C
24 hours at 1-6 C after thawing |
|
|
Product = Cryo
Storage = ? |
1 year at -18 C 6 hours at 20-24 C after thawing (4 hours if pooled) |
|
|
Product = RBCs
QC = ? |
HCT < 80% (all) |
|
|
Product = RBCs leukoreduced
QC = ? |
</= 5 x 10^6 WBCs in 95%, retain 8% of RBCs |
|
|
Product = Platelets
QC = ? |
>/= 5.5 x 10^10 and pH >/= 6.2 in 90% |
|
|
Product = Platelets leukoreduced
QC = ? |
AND < 8.3 x 10^5 WBCs in 95% |
|
|
Product = Apheresis Platelets
QC = ? |
>/= 3.0 x 10^11 and pH >/= 6.2 in 90% |
|
|
Product = Apheresis Platelets leukoreduced
QC = ? |
AND < 5.0 x 10^6 residual WBCs in 95% |
|
|
Product = Cryo
QC = ? |
Factor VIII >/= 80 IU (all) Fibrinogen >/= 150 mg (all) |
|
|
Product = Granulocyte concentration
QC = ? |
>/= 1.0 x 10^10 in 75% |
|
|
Specifics of a RBC unit |
~200 mL RBCs; HCt </= 80% Plasma ~50mL with CPDA-1 WBCs (10^9) & plts Anticoagulant Additive solution (if applicable) 200mg of Iron |
|
|
Specifics of a Platelet unit |
Platelets (>/= 5.5 x 10^10 in 90% tested) ~200 mL plasma (including ~80mg fibrinogen) WBCs (10^7) pH >/= 6.2 (90% tested) |
|
|
Contraindications to Platelet transfusion 1) 2) |
1) TTP (protein deficiency of ADAMTS13) -> leads to large vWF multimers and plt microthrombi -more platelets = more thrombi -HUS similar
2) HIT -Ab vs heparin/platelet factor 4 complex -more platelets = more fuel
3) ITP - sort of, just mostly doesn't help
4) DIC - under discussion |
|
|
ABO and Rh on platelets? |
ABO but NOT Rh antigens present on plts -Doesn't require pre-transfusion crossmatch -May consider Rh ppx with Rh incompatibility (RBCs are present in low quantities) to pre- menopausal D- women
BUT, matched platelets will survive longer |
|
|
Specifics of Apheresis Platelets |
~100mL Platelets (>/= 3 x 10^11 in 90% tested) Plasma (including ~150mg fibrinogen) WBCs (10^6-10^8) <- true source of HLA immunization |
|
|
Leukoreduction, how many WBCs? |
</= 5 x 10^6 WBCs in 95% of tested units Includes RBC, Apheresis, and whole blood units
</= 8.3 x 10^5 WBCs in 95% of test units For platelet concentrate (essentially the same pooled x 6) |
|
|
Why leukoreduce? 6 reasons |
1) Prevention of FHTRs -WBCs secrete pyrogenic cytokines in bag before transfusion (platelets) -Pyrogenic cytokines secreted after transfusion (RBCs)
2) Prevent HLA Immunization -"TRAP" study
3) Prevent CMV transmission -Virus carried only in WBCs (likely monocytes)
4) Prevent immunosuppressive effects
5) Reduction of bacterial contamination
6) Reduction in risk of prion disease - controversial |
|
|
Leukocyte reduction is NOT done to... |
1) Prevent GVHD -Irradiation is only proven method 2) Previously frozen products (leukocytes don't survive) 3) Granulocyte concentrations (haha!) |
|
|
Indications for Washing |
1) Removal of plasma proteins (RBCs & plts) -Classically for IgA deficiency
2) Neonatal alloimmune thrombocytopenic purpura (plts) -Usually due to anti-PL^A1 (HPA-1A); analogous to HDN -Tx with washed maternal plts (lack offending ag & ab)
3) Repeated febrile nonhemolytic reactions -Removes cytokines and WBCs |
|
|
RBC and Platelet shelf-life following washing? |
RBCs = 24 hours Platelets = 4 hours (broken sterility) |
|
|
How does irradiation work? |
1) Deactivates lymphocytes 2) Use to prevent TA-GVHD 3) 2500 cGy ("rad") dose required to target center of bag with at least 1500 cGy in all parts of bag |
|
|
Indications for irradiation? |
1) Immunosuppression -Congenital T-cell deficiencies (DiGeorge, SCID, Wiskott-Aldrich) -Stem cell or marrow transplant recipients -Patients taking Fludarabine 2) Intrauterine and premature neonate txfn 3) Hodgkin disease + heme malignancies 4) Receiving blood from 1st degree relative or "HLA-matched" units |
|
|
Storage after irradiation? |
28 days after irradiation or regular exp date
No change for platelets |
|
|
Specifics of Fresh Frozen Plasma |
Volume 200-250mL All coagulation factors 400mg fibrinogen 1 IU/mL of all others = 100% factor levels Almost no viable cells Anticoagulant |
|
|
FFP Indications |
1) Coagulopathy due to multiple factor deficiencies (liver disease) 2) Urgent reversal of vitamin K deficiency or overdose of warfarin 3) Dilutional coagulopathy 4) Txfn or plasma exchange for TTP/HUS 5) Other factor-specific coagulopathies that don't have factor concentrates |
|
|
Specifics of cryoprecipitate |
~15mL volume >/= 150mg fibrinogen (US ~250mg use for calculation) >/= 80 IU factor VIII 80-120 IU vWF 40-60 IU Factor XIII Fibronectin |
|
|
Indications for Cryoprecipitate |
1) Fibrinogen deficiency (shoot for 100mg/dL) 2) Tx of vWD (2nd line therapy) 3) Tx of uremic thrombocytopathy (dysfibrinogenemia) -1st line DDAVP, dialysis 4) Factor XIII deficiency (factor concentrates) 5) Topical "glue" 6) Tx of Hemophilia A (2nd line) |
|
|
Granulocyte concentrates |
1) Store for 24 hours at 20-24 C without agitation 2) Can irradiate to prevent TA-GVHD - harms lymphs but not really the granulocytes 3) Can't filter to prevent CMV - need CMV- donor 4) Has abundant RBCs so must be ABO & Rh compatible; requires crossmatch |
|
|
DDAVP Functions? |
1) Synthetic form of ADH used for diabetes insipidus 2) Causes release of vWF from endothelial cell storage; functionally increases FVIII |
|
|
DDAVP Indications |
1) Uremic thrombocytopathy 2) Mild Hemophilia A 3) vWD - works in types without marked deficiency -DON'T USE: Type IIB (causes clotting) or III (ineffective)
Effect diminishes with repeated doses |
|
|
Main indicator of survival of an acute HTR? |
Amount of incompatible blood infused |
|
|
Transfusion Reaction Workup should include...? |
1) Clerical check 2) Visible hemoglobinemia check 3) DAT (looks for coated RBCs in vivo) 4) Repeat ABO testing
Not required but often done: 1) Repeat ab screen 2) Indirect bilirubin 3) Haptoglobin (levels DECREASE in acute intravascular hemolysis) 4) Urine hemoglobin |
|
|
Acute Hemolytic Transfusion Reactions |
1) Disastrous, may be fatal 2) #1 cause = clerical error 3) May be intravascular (schistocytes) or extravascular (spherocytes) 4) Most commonly ABO-related 5) Fever & Chills most common presenting sx 6) Abs mostly IgM and great complement-fixers |
|
|
Febrile Nonhemolytic Transfusion Reactions |
1) Most frequently reported reaction 2) Cause: increased pyrogenic cytokines 3) Prevent: leukoreduction |
|
|
Bacterial Contamination |
1) #1 infectious risk from transfusion 2) Red cells: Yersinia, Citrobacter, E. coli, Pseudomonas 3) Platelets: gram + cocci (skin contaminants) |
|
|
TRALI (Transfusion-related acute lung injury) |
1) #1 cause of transfusion-related fatality in US 2) Most common with plts and plasma 3) Hypothesized causes: anti-HLA or anti-neutrophil antibodies from the donor attack recipient white cells; damages capillaries -> leakage and edema 4) Use of male plasma shown to decrease risk of TRALI |
|
|
Hypersensitivity-type reactions |
1) Urticarial: 2nd most frequently reported -Type 1 hypersensitivity to donor plasma proteins -Tx: Benadryl
2) Anaphylactic: classically in IgA deficient recipient -Acute hypotension, abd pain, systemic crash -Tx: washed products or IgA deficient products
|
|
|
Anaphylactoid reactions |
1) Rxns associated with ACE inhibitors -Famous in association with bedside leukoreduction filters -Increased bradykinin (either from contact with filter or ACE inhibitor prevents metabolism)
2) Milder forms of IgA deficiency |
|
|
TACO (Transfusion-associated circulatory overload) |
Acute onset of CHF as a direct result of blood transfusion
Consider lower volume units or volume-reduction |
|
|
Delayed hemolytic transfusion reactions |
Hemolysis occuring days to weeks after transfusion 1) A previously formed ab comes back after re-exposure 2) Typical for Kidd & Duffy 3) Classically extravascular - but delayed HTRs due to Kidd abs may be intravascular and severe 4) DAT classically "mixed field" |
|
|
Post-transfusion Purpura |
1) Thrombocytopenia about 1 week after transfusion 2) Platelets or RBCs can initiate 3) Multiparous females at risk 4) Anti-PL^A1 (HPA-1A) most common (~70%) 5) Transfusion after ab is formed leads to devastating destruction of all platelets 6) Tx: IVIG |
|
|
Viral Disease Risk Transmission |
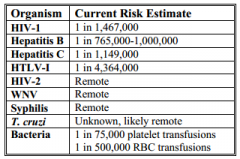
|
|
|
Hepatitis A virus |
1) Fecal-oral transmission 2) Not tested 3) Some concern in pooled products 4) Not prone to chronicity like HBV and esp HCV |
|
|
Hepatitis B virus |
1) DNA virus - hepadnavirus 2) Blood transmission 3) Incubation ~6 weeks 4) Chronic infection "carrier state" much less likely than with HCV (< 5%of adult infections) 5) Currently the most likely virus to be transmitted via transfusion |
|
|
Hepatitis B Viral Serologies |
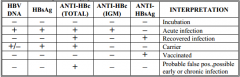
|
|
|
Hepatitis C virus |
1) RNA virus 2) Strong association with chronic hepatitis (75%) 3) Antibody test is anti-HCV (EIA/ChLIA) 4) HCV NAT reduces window period to ~7 days
|
|
|
HIV-1 & HIV-2 Testing |
1) Ab testing required since 1985 2) Window period now down to 10 days 3) HIV NAT 4) Western Blot and IFA for confirmation |
|
|
HTLV-1/II Testing in Transfusion medicine |
1) Transmission through cellular products only 2) HTLV-I disease associations: -Adult T-cell leukemia/lymphoma -HTLV-associated myelopathy (HAM or "tropical spastic paraparesis") 3) HTLV-II no clear-cut associations |
|
|
CMV Virus testing in transfusion medicine |
1) Not required but available 2) Extremely common DNA virus that lives in WBCs only |
|
|
Trypanosoma cruzii (Chagas' disease) testing |
1) Screening test required as of 2011 2) EIA 3) Transmitted through bite of reduviid bug in Central/South America 4) Specific question on donor questionnaire (permanent deferral for hx of Chagas' disease) but many are asymptomatic |
|
|
QC for deglycerolized RBCs? |
Osmolarity > 400 mOsm RBC recovery > 80% Plasma hemoglobin < 300mg/dL |